第二章
贫血
第一节
概述

林果为
【贫血的定义和诊断标准】
贫血(anemia)是指人体循环红细胞容量减少而言。临床上常以外周血单位容积内血红蛋白(Hb)量、红细胞(RBC)数和(或)血细胞比容(Hct)代替红细胞容量来反映贫血程度,一般都以Hb量低于正常参考值95%的下限作为贫血的诊断标准。血红蛋白浓度的降低一般都伴有相应红细胞数量或血细胞比容的减少,但也有不一致。个别轻型缺铁性贫血或珠蛋白生成障碍性贫血可仅有血红蛋白减少,而红细胞数量和血细胞比容都在正常范围内。单位容积血液中血红蛋白量因地区、年龄、性别以及生理性血浆容量的变化而异。婴儿和儿童的血红蛋白量约比成人低15%。男女之间的差异在青春期后才逐渐明显。妊娠时血容量增加,血红蛋白和红细胞数可因被稀释而相对减少。男性65岁以后Hb测定值较65岁以前为低,但女性无差异。国外掌握贫血诊断的Hb标准较统一,都以1972年WHO制订的诊断标准为依据。在海平面地区Hb低于以下水平可诊断为贫血:6个月到6岁儿童110g/L,6~14岁儿童120g/L,成年男性130g/L,成年女性(非妊娠)120g/L,妊娠女性110g/L。而国内诊断贫血的标准都参照下述标准:在海平面地区,成人男性Hb低于120g/L,成年女性低于110g/L,妊娠妇女低于100g/L。是否需要制订老年人贫血诊断标准尚有不同意见,有采用Hb<110g/L作为65岁以后老年人贫血诊断标准,不分男女。选用某一血红蛋白值来划分有无贫血,要做到非常合理是相当困难的。因为正常人群血红蛋白分布曲线和贫血人群血红蛋白分布曲线之间互有重叠。事实上Hb正常值的个体差异较大,如某患者一周前Hb 155g/L,现Hb降低为140g/L,虽然在正常范围,但应认为是有意义的。决定患者是否有贫血时尚须注意Hb测定的标准化,以及采血的部位,指端血、耳垂血、静脉血其测定值可略有不同。WHO规定的标准方法为静脉血氰化高铁Hb法。此外,血浆容量的生理和病理变化,如妊娠后3个月、全身水肿、充血性心力衰竭、低蛋白血症以及某些细胞因子的作用,因血浆容量增加血液被稀释,Hb量下降,可误认为贫血,也称为稀释性假性贫血;血浆容量的丢失如失水、腹泻、呕吐、重度烧伤或大量使用利尿剂后血液浓缩,Hb量可上升,即使有贫血检测值也可正常。急性大量失血,红细胞和血浆同时丢失,虽然红细胞丢失过多,但贫血可不明显。贫血按严重程度可分为:极重度贫血,Hb≤30g/L;重度贫血,Hb 31~60g/L;中度贫血,Hb 61~90g/L;轻度贫血,Hb>90g/L与低于正常参考值的下限之间。
贫血是一种症状,而不是具体的疾病。各种疾病都可伴有贫血。如果许多原因不同的贫血具有类似的临床表现和血液学特征,则可归纳为一种综合病征,如再生障碍性贫血、缺铁性贫血等。贫血在世界各地属常见病,在发展中国家以及血红蛋白病或葡萄糖-6-磷酸脱氢酶变异的多民族及地区,贫血问题尤为突出。
【发病机制】
(一)红细胞生成减少
骨髓造血活动与造血组织中造血干细胞的存在有密切的关系。造血干细胞在特定的微环境下分化成各系列祖细胞,经各系前体细胞发育成各系成熟细胞。当某些化学、物理、病毒感染和免疫因素损伤造血干细胞和(或)造血微环境,致使造血干细胞数量减少或质的异常致分化、增殖发生障碍,导致骨髓造血衰竭、周围血液全血细胞减少,称为再生障碍性贫血(aplastic anemia)。遗传因素也可引起骨髓造血衰竭。
造血干细胞在造血微环境诱导下分化为红系祖细胞,后者在红细胞生成素(EPO)的刺激下分化为各期幼红细胞。红系祖细胞或红细胞生成素的免疫性破坏,或红系祖细胞受病毒(人类微小病毒,HPV-B19)感染和溶解,均可导致选择性红系细胞生成障碍。贫血严重而白细胞和血小板大致正常,称为纯红细胞再生障碍性贫血(pure red cell aplasia)。EPO产生不足和红系祖细胞对EPO反应迟钝是肾性贫血(renal anemia)和慢性病贫血(anemia of chronic disease)的主要发病机制之一。
自红系祖细胞发育至中幼红细胞,细胞要经过多次分裂增殖,而DNA的合成倍增是细胞分裂期前所必需的。维生素B 12 和叶酸则是DNA合成的主要辅酶。无论是维生素B 12 或叶酸缺乏或由于其他因素影响DNA合成,都可导致核分裂延迟甚至停顿;形成核和胞质发育不平衡、核染色质疏松、形态巨大而畸形的巨幼红细胞。周围血液可见卵圆形的大红细胞,称为巨幼细胞贫血(megaloblastic anemia)。
在幼红细胞不断增殖的过程中,细胞质也逐渐发育成熟。早在早幼红细胞胞质内就开始合成微量血红蛋白,至中幼红细胞阶段血红蛋白合成达到高峰,一直持续到网织红细胞。血红蛋白的合成需要铁。铁通过血浆中的运铁蛋白运输到幼红细胞表面,和幼红细胞表面的运铁蛋白受体结合,通过胞饮方式进入质内,输送到线粒体,和原卟啉合成正铁血红素。珠蛋白是在幼红细胞内的核糖体上合成的。正铁血红素与珠蛋白合成血红蛋白分子。所以任何原因引起的血红蛋白合成障碍,不论是缺铁(缺铁性贫血)或铁代谢紊乱(慢性病贫血)、珠蛋白合成障碍(血红蛋白病)以及血红素卟啉环合成障碍(铁粒幼细胞性贫血)等,都可以导致Hb合成障碍,出现大量细胞质不足(小红细胞)及血红蛋白含量减少(低色素)的成熟红细胞,统称为低色素性贫血,其中以缺铁性贫血最常见。
骨髓发生纤维化或骨髓被异常细胞所侵犯,可导致骨髓结构和功能的破坏,同时伴有骨髓外造血灶的建立。临床上出现贫血,周围血液出现幼粒和幼红细胞,称为幼粒-幼红细胞贫血或骨髓病性贫血(myelophthisic anemia)。
无效红细胞生成是指患者骨髓增生,幼红细胞增多,但由于幼红细胞本身有缺陷导致过早在骨髓凋亡,引起红细胞生成减少,网织红细胞减少,导致贫血。见于骨髓增生异常综合征难治性贫血、巨幼细胞贫血及珠蛋白生成障碍性贫血等。
(二)红细胞破坏过多
红细胞破坏过多引起的贫血,称溶血性贫血(hemolytic anemia),是由于红细胞破坏增加(寿命缩短),超过骨髓造血代偿能力时而发生的贫血。骨髓造血具有产生红细胞6~8倍的造血代偿潜力,如果红细胞破坏速率在骨髓造血的代偿范围内,则虽然有溶血,红细胞破坏,但不出现贫血,称为溶血性疾患。正常红细胞的寿命约120天,只有在红细胞的寿命缩短至低于15~20天,红细胞破坏速度超过骨髓造血的代偿潜力时才会发生贫血。溶血性疾患有黄疸表现者称溶血性黄疸,黄疸的有无取决于溶血程度和肝脏处理胆红素的能力,因此溶血性贫血不一定都有黄疸。
溶血性贫血的根本原因是红细胞寿命缩短,易于破坏。造成红细胞破坏加速的机制可概括为红细胞本身的内在缺陷和红细胞外部因素异常。前者多为遗传性溶血,后者引起获得性溶血。红细胞内在缺陷包括红细胞膜缺陷、红细胞酶的缺陷和血红蛋白异常。红细胞膜缺陷多因基因突变致红细胞膜骨架蛋白异常,引起红细胞形态改变,这种形态异常红细胞容易在单核-巨噬细胞系统内破坏,如遗传性球形红细胞增多症,也可因造血干细胞克隆性病变引起获得性红细胞膜缺陷,受累红细胞对补体介导的溶血敏感性增高,造成血管内溶血称阵发性睡眠性血红蛋白尿。参与红细胞代谢的酶(糖代谢酶)由于基因突变使酶活性改变,导致无氧糖酵解途径酶缺陷可造成红细胞能量来源不足,使细胞膜功能异常,产生溶血,如丙酮酸激酶缺乏症。磷酸戊糖旁路代谢酶缺陷的结果造成还原型谷胱甘肽的减少,细胞易受氧化损伤而发生溶血,如葡糖糖-6-磷酸脱氢酶缺乏。因基因突变,使珠蛋白肽链结构异常(异常血红蛋白病)或肽链合成异常(珠蛋白生成障碍性贫血),导致红细胞硬度增加。或异常血红蛋白在红细胞内形成聚合体、结晶体或包涵体,造成红细胞变形性降低,通过单核-巨噬细胞系统特别是脾时,破坏增加。
红细胞外在因素引起溶血性贫血都为获得性,有免疫性因素和非免疫性因素两种。免疫性溶血是抗原抗体介导的红细胞破坏。自身免疫性溶血性贫血患者产生抗红细胞抗体,温抗体型为不完全抗体,与红细胞结合后,致敏红细胞在单核-巨噬细胞系统内被破坏或清除,是免疫性溶血性贫血中最常见的类型。冷抗体型多为完全抗体,可使红细胞直接在血管内破坏。血型不合输血亦可造成血管内溶血。新生儿溶血病是因为母婴血型不合,母亲产生的抗胎儿血型IgG型抗体通过胎盘进入胎儿血液循环,造成溶血,最常见的是ABO血型不合,其次是Rh血型不合。非免疫因素包括各种感染(如疟疾等)、某些化学物质(包括药物)和毒物可以通过氧化或非氧化作用破坏红细胞。葡萄糖-6-磷酸脱氢酶缺乏症患者对氧化性物质特别敏感。药物性溶血性贫血分为药物诱发免疫性和非免疫性溶血性贫血两种。物理和创伤性因素包括人工心脏瓣膜可以引起红细胞的机械性破坏;微血管病性溶血性贫血是因为微血管内皮损伤或纤维蛋白网络形成,红细胞在通过狭窄的血管腔时,造成红细胞破坏,见于弥散性血管内凝血、溶血性尿毒症综合征和血栓性血小板减少性紫癜;行军性血红蛋白尿症是敏感个体因行军和赛跑而造成的红细胞机械性破坏;烧伤可直接破坏红细胞。生物毒素引起溶血,以蛇毒最常见。
(三)红细胞丢失过多
不论急性或慢性失血都是临床上引起贫血最常见的原因。慢性失血性贫血实质上就是缺铁性贫血。
贫血的发病机制往往是多因素的。例如恶性肿瘤所致贫血的发生机制有失血(失血性贫血)、骨髓浸润(骨髓病性贫血)、肿瘤广泛转移在微血管形成瘤细胞栓(微血管病性溶血性贫血)、营养障碍致造血物质缺乏(营养性贫血)、红细胞生成素减少(慢性病贫血)、化疗和放疗的应用(治疗相关性贫血)。此外,某些肿瘤如胸腺瘤患者体内可产生抗幼红细胞或抗EPO抗体,致纯红细胞再生障碍性贫血,淋巴瘤等可导致自身免疫性溶血性贫血,多发性骨髓瘤等因血浆球蛋白异常增多,大量细胞外液进入血管内可致稀释性贫血。药物也能通过不同机制引起多种类型的贫血,许多药物可抑制骨髓造血引起再生障碍性贫血(如抗肿瘤药物和氯霉素等),某些药物可影响红系细胞的DNA合成,引起巨幼细胞贫血(如抗代谢药、抗癫痫药等),阿司匹林可引起胃肠道出血致缺铁性贫血,抗结核药可引起铁粒幼细胞性贫血,药物或其代谢产物可与红细胞膜发生作用,导致新抗原形成,引起药物免疫性溶血性贫血,如奎尼丁、非那西丁、磺胺药等,药物还能作用于有遗传性酶缺陷或异常血红蛋白的患者,引起溶血性贫血发作。同一类型的贫血也可有多种发病机制并存,如巨幼细胞贫血既有DNA合成障碍,又有红细胞破坏过多和幼红细胞过早在髓内凋亡等因素。
【分类】
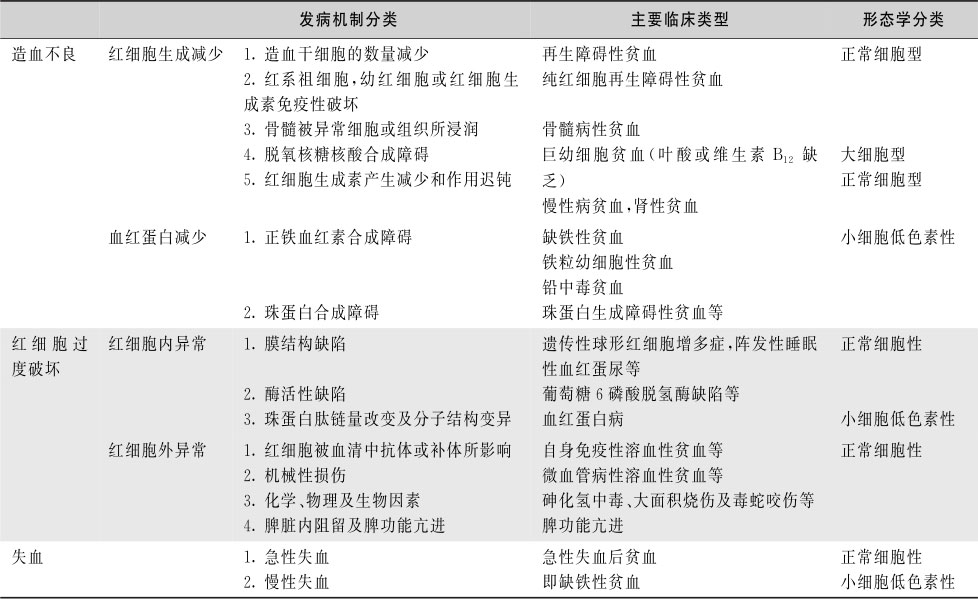
(一)贫血的形态学分类
贫血可按不同的发病机制和细胞形态学特征进行分类(表16-2-1)。按发病机制可分为造血不良、红细胞过度破坏及急、慢性失血三类。按形态学分类,则可分为正常细胞性、大细胞性和小细胞低色素性三类。形态学分类不是固定不变的,例如再生障碍性贫血多数是正常细胞性贫血,但偶可呈大细胞性贫血;溶血性贫血和急性失血后贫血也可呈正常细胞性贫血也可呈大细胞性贫血。贫血的形态学分类虽过于简单,但易于掌握,可提供诊断线索,如小细胞低色素性贫血多数是缺铁性贫血,大细胞性贫血很可能是由维生素B 12 或叶酸缺乏所引起。
(二)溶血性贫血的分类和临床表现
按病情可分为急性和慢性溶血性贫血。按溶血的场所可分为血管内溶血和血管外溶血。按病因可分为遗传性和获得性溶血性贫血。按发病机制可分为红细胞内异常和红细胞外异常引起的溶血性贫血。
表16-2-1 贫血的发病机制和形态学分类
临床表现:①急性溶血:急性溶血性贫血起病急骤,短期大量溶血引起寒战、高热、头痛、呕吐、四肢腰背疼痛,紧接着出现血红蛋白尿,其后出现黄疸。由于红细胞大量破坏,其分解的产物对机体产生毒性作用,严重者可发生周围循环衰竭。红细胞破坏的产物可引起肾小管坏死和管腔阻塞,导致急性肾衰竭。②慢性溶血:慢性溶血性贫血多为血管外溶血,发病缓慢,表现为贫血、黄疸和脾大三大特征。长期的高胆红素血症,可并发胆石症和肝功能损害。③血管内溶血:以急性溶血多见,多有腰背酸痛、高热并伴有血红蛋白血症、血红蛋白尿。也有慢性血管内溶血,可有含铁血黄素尿。见于阵发性睡眠性血红蛋白尿、红细胞破碎综合征、ABO血型不合所致输血反应、阵发性冷性血红蛋白尿、部分感染(如恶性疟疾、梭状芽孢杆菌败血症)、化学因子(砷、蛇毒、蜘蛛毒)引起的溶血性贫血、输注低渗溶液及热损伤引起的溶血性贫血。④血管外溶血:血管外溶血主要发生于脾,临床表现一般较轻,可有血清游离胆红素轻度升高,一般不出现血红蛋白尿,可有脾大。
【病理生理和贫血的一般临床表现】
贫血的病理生理学基础是血红蛋白减少,血液携氧能力减低,全身组织和器官发生缺氧变化等。首先体内相应的代偿机制发挥作用,例如脉搏变快、心搏输出量增加、呼吸加速、红细胞生成素分泌增多,以及血红蛋白与氧的亲和力降低等。有些脏器(如肾脏等)则发生血管收缩,使更多的血液流向缺氧较为敏感的器官如脑、心脏等。红细胞内合成更多的2,3-二磷酸甘油酸(2,3-DPG),后者与脱氧血红蛋白的β链相结合,以降低血红蛋白对氧的亲和力,血红蛋白氧解离曲线右移,使组织获得更多的氧。轻、中度贫血患者持续一定时期后,可由于这种代偿机制而不表现明显的缺氧症状。
贫血症状的有无及轻重,除原发疾病的性质外,主要取决于贫血的程度及其发生的速度,同时也与病人年龄、有无其他心肺疾病以及心血管系统的代偿能力有关。慢性贫血,无心肺疾病基础,代偿机制可充分发挥,即使血红蛋白低达80g/L亦可无症状;有时低至60g/L以下才引起病人的注意。反之,急性溶血和急性失血,虽然贫血不很严重,但由于发生较快来不及代偿,症状却很显著。儿童及年轻患者由于其心血管系统代偿功能良好,往往较年老患者容易耐受贫血的影响。
(一)一般表现
皮内毛细血管缺血所致的皮肤黏膜苍白,是贫血最常见的体征。但影响皮肤颜色的因素很多,除血红蛋白量外,还和皮内毛细血管分布和舒缩程度、皮肤色素和皮下组织含水量的多寡有关。因此单凭皮肤颜色判断贫血程度常有偏差,一般以观察指甲、手掌皮肤皱纹处以及口唇黏膜和睑结膜等较为可靠。疲倦、乏力、头晕耳鸣、记忆力衰退、思想不集中等都是贫血早期和常见的症状,可能由于神经系统及肌肉缺氧所致。贫血严重时可有低热和基础代谢率增高。
(二)呼吸系统
稍事活动或情绪激动即有气急。由于血红蛋白量减少,活动增加必然引起血氧含量进一步降低和二氧化碳含量增高,反射性地刺激呼吸中枢,发生呼吸急促。
(三)循环系统
中度贫血患者常表现为窦性心动过速、心搏亢进、脉搏充实、脉压增宽、循环时间加速及心排出量增多等。肺动脉瓣或心尖区可听到中等响度的吹风样收缩期杂音,其产生原因与血循环加速、血粘度以及缺氧后心肌张力降低有关。当心脏扩大时,杂音还可因二尖瓣和三尖瓣相对性关闭不全所致。当血红蛋白量低于60g/L时,约30%患者可有心电图改变,表现为低电压、ST段压低、T波平坦倒置,严重者甚至可有QT时间延长、心房颤动等。发生心律失常,要考虑是否合并有其他心脏疾患。严重贫血(血红蛋白低于30g/L以下)或贫血进展较速的病例,可有明显的全心扩大;以后由于心肌营养障碍,无法代偿日益增加的高输出量状态,最终导致充血性心力衰竭。当贫血被纠正后,上述心脏病变可获得一定程度的恢复。重度贫血患者即使无充血性心力衰竭,但由于血清白蛋白减少、毛细血管通透性增加以及肾血流量减少,引起水、钠潴留,可发生水肿。
(四)消化系统
贫血影响消化系统的功能和消化酶的分泌,出现食欲减退、恶心、呕吐、腹胀甚至腹泻。部分患者有明显的舌炎。消化系统表现除因贫血缺氧外,还与原发疾病有关。
(五)泌尿生殖系统
贫血时肾血管收缩和肾脏缺氧,可导致肾功能变化。早期有多尿、尿比重降低及血尿素氮增多,贫血严重时可出现蛋白尿。月经失调(闭经)和性欲减退也颇常见。
【实验室检查】
实验室检查既是确立贫血的可靠方法,又是明确其类型的重要步骤。兹将贫血的常规实验室检查项目简述如下。各类贫血的特殊检查见各病。
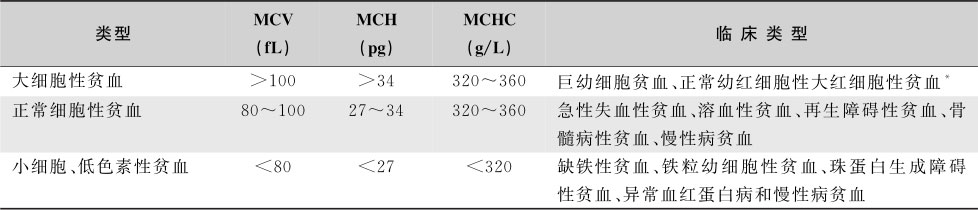
(一)红细胞参数
根据自动血细胞分析仪获得的参数,有助于贫血的形态学分类(表16-2-2)。其中以MCV和RDW意义最大,MCV是反映红细胞大小的参数,RDW是反映红细胞大小不一程度的参数。
表16-2-2 贫血的细胞形态学分类
注:* 包括溶血性贫血、急性失血后贫血、肝病贫血、难治性贫血
(二)周围血涂片
周围血涂片检查对贫血的诊断具有重要价值,应视为必检项目。它不仅有助于贫血的形态学分类,而且又能从中发现异形红细胞。红细胞有大小不均、小型红细胞增多且中央苍白区扩大,可诊断为低色素性贫血(hypochromic anemia)。球形红细胞增多,见于遗传性球形红 细胞增多症(hereditary spherocytosis)和自身免疫性溶血性贫血(autoimmune hemolytic anemia,AIHA),椭圆形红细胞增多见于遗传性椭圆形红细胞增多症(hereditary elliptocytosis)等,镰形红细胞见于镰状细胞贫血(sickle cell anemia),口形红细胞见于遗传性口形红细胞增多症(hereditary stomatocytosis)等,棘形红细胞见于先天性无β脂蛋白血症和肝衰竭,靶形红细胞常见于珠蛋白生成障碍性贫血(thalassemia)。各种异形红细胞,如梨形、哑铃形、三角形甚至红细胞碎片,则提示微血管病性溶血性贫血(microangiopathic hemolytic anemia)的可能性。泪滴状红细胞可见于骨髓纤维化。出现幼粒幼红细胞是骨髓病性贫血(myelophthisic anemia)的重要依据。溶血性贫血周围血液可出现幼红细胞,但数量不多,主要是晚幼红细胞,严重溶血时数量增多且可见豪-胶小体和幼粒细胞,由于网织红细胞和其他较不成熟红细胞自骨髓中大量释放到血液,故周围血液中大红细胞增多。
(三)网织红细胞计数
网织红细胞(reticulocyte,Ret)是晚幼红细胞脱核后尚未完全成熟的红细胞,因细胞质内残存核糖体RNA,经煌焦油蓝或新亚甲蓝活体染色后在显微镜下呈网状而得名。Ret在骨髓停留2~3天,外周血停留1天后变为成熟红细胞。显微镜下人工计数因影响因素较多,当Ret较低时,误差较大。血细胞自动分析仪计数,精确度比人工计数提高了10倍以上。新型的血细胞自动分析仪采用流式激光技术和荧光染色技术测量Ret胞质内的RNA含量,并根据荧光强度,将Ret分为高荧光强度Ret(HFR)、中荧光强度 Ret(MFR)和低荧光强度 Ret(LFR),并计算未成熟 Ret分数(IRF)[IRF=(HFR+MFR)/Ret]。IRF值与RNA含量和Ret幼稚程度呈正相关,能更好反映骨髓红系的造血功能,因此IRF是评估Ret成熟度的参考指标,其正常参考值为5%~22%。
当外周血有幼红细出现时要影响分析仪计数的测定值,故该时应推荐手工测定。
网织红细胞计数是反映骨髓红系增生情况的重要指标,是临床上鉴别红系增生不良性贫血和溶血性贫血最简单的方法。溶血性贫血及急性失血性贫血,其贫血原因系外周性,骨髓代偿性增生功能良好,故网织红细胞数增高,溶血性贫血一般可高达5%~20%,但溶血性再障危象,网织红细胞数可减少;再生障碍性贫血和纯红细胞再生障碍性贫血网织红细胞常有显著减少,骨髓幼红细胞无效增生即髓内过早凋亡,网织红细胞也减少,因此网织红细胞减少常提示贫血原因是骨髓性的。
由于网织红细胞的百分数受红细胞总数的影响,当外周血红细胞数减少时,可使网织红细胞百分数增高,但实际上从骨髓释放的网织红细胞并无增多,因此必须计算绝对值才能反映真实情况。
网织红细胞绝对值(×10 9 /L)=[网织红细胞(%)×红细胞数(10 12 /L)×10 12 ]/100
临床应用网织红细胞参数应同时报告百分数、绝对值及IRF。切记无贫血时Ret绝对值的参考值为(25~75)×10 9 /L,有贫血Ret绝对值<75×10 9 /L表示贫血是低增生的或红系有成熟障碍;诊断纯红细胞再生障碍性贫血,网织红细胞绝对值要求低于10×10 9 /L;如网织红细胞绝对值>100×10 9 /L常提示贫血的性质为溶血性或出血性及营养性贫血补充治疗后红系的增生反应。
(四)骨髓象检查
根据骨髓增生与否,可将贫血分为增生性和增生不良性两大类。再生障碍性贫血属骨髓增生不良;缺铁性贫血、巨幼细胞贫血、溶血性贫血、急性失血性贫血骨髓增生良好,骨髓增生异常综合征贫血的骨髓增生多数也是良好的;单纯幼红细胞减少或缺如应疑及纯红细胞再生障碍性贫血或继发于肾脏及内分泌疾病的贫血。骨髓涂片检查是确定巨幼细胞贫血的重要方法,但出现幼红细胞巨幼样改变尚见于骨髓增生异常综合征贫血及红白血病等。骨髓涂片铁染色检查是诊断缺铁性贫血和铁粒幼细胞性贫血的重要依据,慢性病贫血有铁利用障碍(骨髓小粒可染铁增多,但铁粒幼细胞减少)。骨髓“干抽”常是诊断骨髓纤维化贫血的线索之一。
(五)溶血性贫血的一般实验室检查
溶血性贫血一般实验室检査的任务是确定患者是否存在溶血。临床常用的检查有:
1.有关溶血性黄疸的检查
①血清非结合胆红素水平增加:大量溶血时,血清非结合胆红素增加,结合胆红素常少于总胆红素的15%。由于肝脏清除胆红素的能力很强,黄疸仅轻度或中度,即使大量溶血时,一般也不超过85.5μmol/L。血清胆红素浓度除了取决于血红蛋白分解的程度外,还与肝脏清除胆红素的能力密切相关。慢性溶血性贫血患者长期高胆红素血症,会引起肝功能损害,合并肝细胞性黄疸。②尿胆原增加:正常人每天从尿中排出的尿胆原为0~5.9μmol/L。急性大量溶血时,尿胆原排出量明显增多。慢性溶血时,只有当肝功能减退时,尿胆原才会增加。③粪胆原排出增加:正常人每天从粪便中排出的粪胆原为68~473μmol/L。当血红蛋白大量分解时,每日粪胆原排出量可多达680~1700μmol/L,甚至可高达2550μmol/L。
2.有关血管内溶血的检查
①血红蛋白血症:正常血浆只有微量的游离血红蛋白,含量1~10mg/L。当大量溶血时,主要是血管内溶血时可高达1000mg/L以上。由于血液标本在体外储存时容易造成溶血,游离血红蛋白检测的实际意义不大。②血红蛋白尿:血浆中的游离血红蛋白超过了结合珠蛋白所能结合的量时,多余的血红蛋白即可从肾小球滤出。血红蛋白尿需和肌红蛋白尿进行鉴别,肌红蛋白是小分子片段,极容易经肾过滤,不会在血浆中积蓄变成肉眼所见的红色,因此观察到血浆变成红色可有助于血红蛋白尿的确定。③含铁血黄素尿:被肾小管重吸收的游离血红蛋白,在近曲小管上皮细胞内被分解为卟啉、铁和珠蛋白。铁以含铁血黄素形式沉积在上皮细胞内,当细胞脱落随尿排出,即成为含铁血黄素尿。含铁血黄素尿主要见于慢性血管内溶血,急性血管内溶血时,含铁血黄素尿要几天后才阳性,并可持续一段时间。④血清结合珠蛋白降低:血清结合珠蛋白是血浆中的一组α 2 糖蛋白,是血红蛋白的转运蛋白,在肝内产生,正常血淸含量为500~1500mg/L。当血管内溶血后,1分子的结合珠蛋白可结合1分子的游离血红蛋白。此种结合体很快地从血中被肝细胞清除。血清结合珠蛋白的降低可见于血管内溶血,亦可见于血管外溶血,特别是在微血管病性溶血性贫血时是最敏感的溶血指标,可出现在贫血或血红蛋白血症之前。血清结合珠蛋白降低也见于巨幼细胞贫血、髓内溶血和肝病时,而在感染及恶性肿瘤中可升高。
3.血清乳酸脱氢酶(LDH)
溶血性贫血时红细胞内酶(LDH 1 、LDH 2 、LDH 3 )大量进入血浆所致。但很多原因可以引起LDH增高,因此缺乏特异性。
4.红细胞寿命缩短
红细胞寿命测定为诊断溶血的可靠指标。常用 51 Cr、 32 P-DFP或 3 H-DFP(二异丙基氟磷酸)标记红细胞。 51 Cr仅代表红细胞寿命指数,后两者测定比较接近红细胞的寿命且敏感。红细胞肌酸是红细胞寿命的定量指标,因为年轻红细胞肌酸水平高于年老的红细胞,因此可作为溶血的指标,特别是血管内溶血。
【贫血的诊断步骤和思路】
贫血的诊断一般分三个步骤:①贫血及其严重度的确立;②贫血的性质诊断,即属于何种贫血综合征;③贫血的病因诊断。
贫血的性质诊断可以从贫血的形态学分类入手。小细胞低色素性贫血以缺铁性贫血最常见,因此可从铁代谢检查入手(图16-2-1);如属铁利用障碍,要考虑慢性病贫血,如属体内铁过多,则可能是铁粒幼细胞性贫血或血红蛋白病,前者经骨髓涂片铁染色即可确诊,后者需要借助血红蛋白性质的检测。大细胞性贫血经骨髓检查可区分巨幼细胞贫血和非巨幼细胞大细胞性贫血,后者见于急性失血性贫血、溶血性贫血、肝脏病贫血和内分泌功能减退性贫血,通过网织红细胞、血清维生素B 12 、叶酸及相关检查即可确定(图16-2-2)。正常细胞性贫血如有网织红细胞增高,除外急性出血性贫血后,主要是一组溶血性贫血综合征,由于后者最常见的是自身免疫性溶血性贫血,因此可从血清抗人球蛋白试验入手进行鉴别诊断;如网织红细胞不增多,多系骨髓造血过程有障碍所致的贫血,应仔细检查骨髓象(图16-2-3)。
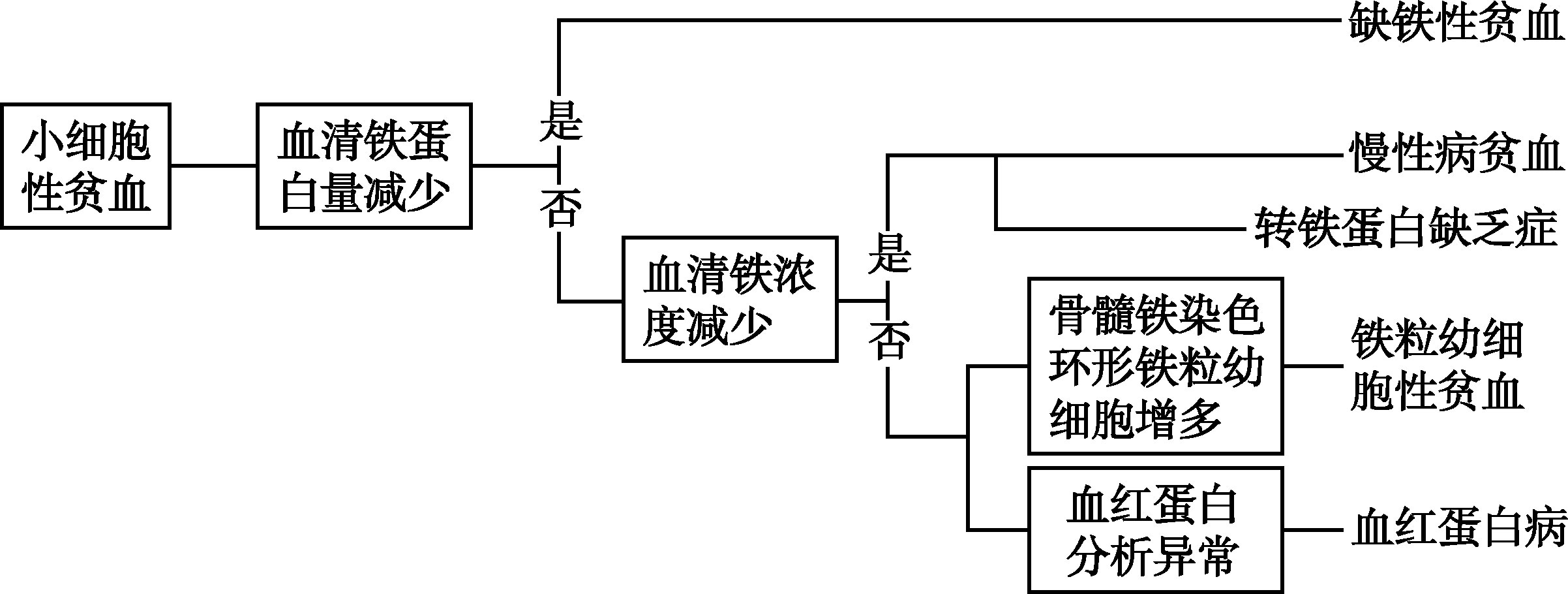
图16-2-1 小细胞性贫血诊断流程
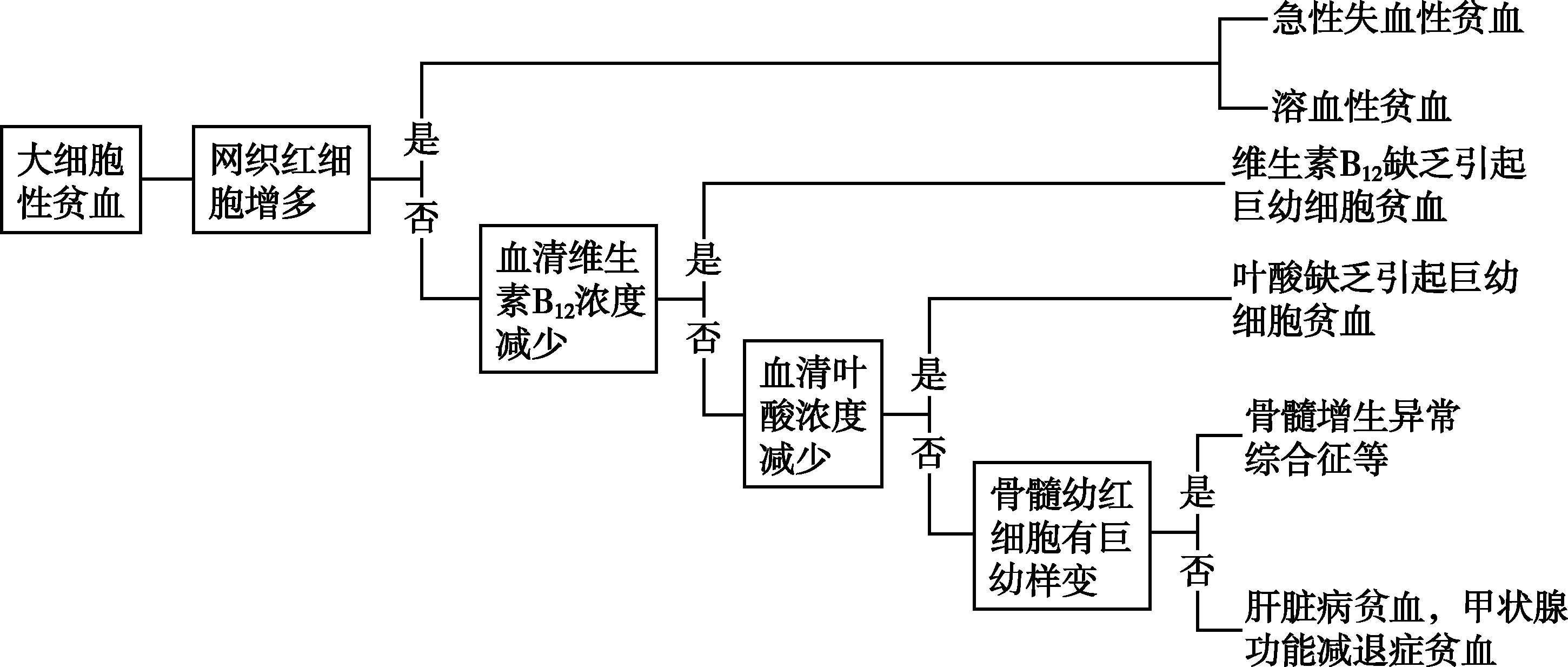
图16-2-2 大细胞性贫血诊断流程

图16-2-3 正常细胞贫血诊断流程
伴有网织红细胞增高或伴有黄疸的贫血不一定都是溶血性贫血,需要注意鉴别诊断。贫血伴有网织红细胞增多尚可见于缺铁性贫血或巨幼细胞贫血补充铁剂、维生素B 12 或叶酸治疗有效时及失血性贫血时。无效造血(骨髓内溶血),周围血可出现幼红细胞,骨髓幼红细胞显著增生。体腔或组织内出血,也可出现贫血伴有无胆红素尿性黄疸,血清非结合胆红素增高。无胆红素尿性黄疸而无贫血,要和以非结合胆红素升高为主的家族性非溶血性黄疸(Gilbert综合征和Crigler-Najjar综合征)、新生儿高胆红素血症等鉴别。
贫血的病因诊断十分重要,目的是不要延误重要疾病特别是恶性肿瘤的诊断。贫血的病因诊断必须从详细询问病史、全面体格检查入手。短期内血红蛋白过速下降应疑为急性失血和急性溶血性贫血,许多遗传性贫血患者常自幼即有贫血史,月经期妇女以缺铁性贫血最常见,成年男性发生缺铁性贫血要高度怀疑胃肠道出血,老年人贫血特别是有有害物质暴露史者应高度怀疑骨髓增生异常综合征贫血。药物暴露史的询问也很重要,药物可致多种类型的贫血。不明原因的贫血患者大便潜血、肾功能检查、已婚妇女的妇科检查、农村来的病人的寄生虫检查等,均应视为常规检查。
主要参考文献
1.张之南,沈悌.血液病诊断及疗效标准.第3版.北京:科学出版社,2007:1-4.
2.林果为,欧阳仁荣,陈珊珊,等.现代临床血液病学.上海:复旦大学出版社,2013:279-290.
3.王小钦,林果为.重新认识网织红细胞参数的临床价值.中华血液学杂志,2014,35(1):1-3.
第二节
再生障碍性贫血和其他骨髓衰竭综合征
林果为
一、再生障碍性贫血
再生障碍性贫血(简称再障,aplastic anemia,AA),是一组最常见的获得性骨髓造血功能衰竭症(bone marrow failure),导致骨髓造血干/祖细胞和三系血细胞产生减少,外周血呈全血细胞减少,但骨髓中无恶性细胞浸润,无广泛网硬蛋白纤维增生。据国内21省(市)自治区的调查,年发病率为0.74/10万。西方国家为0.20/10万。各年龄组均可发病,发病年龄有两个高峰:15~25岁和60~65岁。
【病因和类型】
(一)自身免疫性再障
绝大多数临床诊断原发性的获得性再障是属于自身免疫性疾病,其靶器官为骨髓,最终引起骨髓衰竭。获得性再障应用抗淋巴细胞球蛋白和(或)环孢素等免疫抑制剂治疗后,至少有50%~80%的患者获得缓解;患者骨髓祖细胞体外培养去除T淋巴细胞可使集落生长恢复;再障骨髓寡克隆T细胞内及患者血清中均可检出含高浓度IFN-γ和TNF-α。由于骨髓中IFN-γ和TNF-α产生过多,诱导骨髓CD34 + 细胞大量凋亡,从而引起造血干/祖细胞减少。自身寡克隆抑制性T淋巴细胞产生的机制尚不清楚,可能和调节性T细胞(CD4 + 、CD25 + 和FoxP3 + )功能丧失有关。此外再障可继发于胸腺瘤、系统性红斑狼疮、嗜酸性筋膜炎和类风湿关节炎等,患者血清中可找到抑制造血干细胞的抗体。药物的特异质反应及病毒性肝炎相关性再障也是自身免疫性再障。
(二)药物性再障
有两种类型:①和剂量有关,系药物毒性作用,达到一定剂量就会引起骨髓抑制,如各种抗肿瘤药。其中细胞周期特异性药物主要作用于容易分裂的细胞,因此发生全血细胞减少时骨髓仍保留一定量的多能干细胞,停药后再障可以恢复;白消安和亚硝脲类不仅作用于进入增殖周期的细胞,而且也作用于非增殖周期的细胞,常导致长期骨髓抑制难以恢复。此外,无机砷、雌激素、苯妥英钠、吩噻嗪、硫尿嘧啶及氯霉素等也可以引起与剂量有关的骨髓抑制。②和剂量关系不大,仅个别患者发生造血障碍,多系药物的特异质反应,是自身免疫性的,常导致持续性再障。常见的有氯(合)霉素、有机砷、米帕林、三甲双酮、保泰松、金制剂、氨基比林、吡罗昔康(炎痛喜康)、磺胺、甲砜霉素、卡比马唑(甲亢平)、甲巯咪唑(他巴唑)、氯磺丙脲等。最常见是由氯霉素引起的,氯(合)霉素的化学结构含有一个硝基苯环,其骨髓毒性作用与亚硝基-氯霉素有关,它可抑制骨髓细胞内线粒体DNA聚合酶,导致DNA及蛋白质合成减少,也可抑制血红素的合成,幼红细胞质内可出现空泡及铁粒幼细胞增多。
(三)病毒性肝炎相关性再障
简称肝炎相关性再障(hepatitis associated aplastic anemia,HAAA),是病毒性肝炎最严重的并发症之一,发生率不到1.0%,占再障患者的3.2%。80%病例引起再障的病毒性肝炎亚型至今尚未明确(非甲、乙、丙、丁、戊),但20%病例明确由乙型肝炎引起。临床上有两种类型:急性型居多数,起病急,肝炎和再障发病间期平均10周左右,肝炎已处于恢复期,但再障病情重,生存期短,发病年龄轻,大多病毒性肝炎亚型不明确;慢性型属少数,大多在慢性乙型肝炎基础上发病,病情轻,肝炎和再障发病间期长,生存期也长。肝炎病毒对造血干细胞有直接抑制作用,也可通过病毒介导的自身免疫异常,尚可破坏骨髓微循环。其他病毒如人类微小病毒B19、EB病毒等也有报道。
(四)苯中毒所致再障
苯及其衍化物和再障的关系已为许多实验研究所肯定,苯进入人体易固定于富含脂肪的组织,慢性苯中毒时苯主要固定于骨髓,苯的骨髓毒性作用与其代谢产物(苯二酚、邻苯二酚)有关,酚类为原浆毒,可直接抑制细胞核分裂,所形成的半抗原可刺激免疫反应。
(五)造血干/祖细胞自身缺陷
阵发性睡眠性血红蛋白尿(paroxysmal nocturnal hemoglobinuria,PNH)和再障的关系相当密切,PNH系获得性造血干/祖细胞自身缺陷引起造血衰竭(参见本章第三节“阵发性睡眠性血红蛋白尿”),约30%PNH患者有再障病史,再障患者采用流式细胞术检测PNH克隆阳性率可达25%~67%,甚至临床上有AA-PNH综合征,两者可先后或同时发生。再障患者出现PNH克隆的机制仍不清楚,可能和再障患者造血干/祖细胞“逃逸”免疫攻击而自身选择的结果。近年研究还发现某些获得性再障患者白细胞染色体端粒长度缩短,这些患者常对免疫抑制剂无效。
(六)其他因素
包括:①电离辐射:X线、γ线或中子可直接损害造血干细胞和骨髓微环境。长期超允许量放射线照射(如放射源事故)可致再障。全身照射超过700~1000c Gy可致持久性再障,>4000cGy骨髓微环境被破坏,骨髓不能支持造血。②妊娠:罕有病例报告,再障在妊娠期发病,分娩或人工流产后缓解,第二次妊娠时再发,是否与妊娠激活了免疫反应有关。
【临床表现】
再障可按严重度不同分为严重型、极严重型和非严重型。严重型再障(severe aplastic anemia,SAA)的诊断标准(Camitta标准):①骨髓细胞增生程度<正常的25%;如≥正常的25%但<50%,则残存的造血细胞应<30%。②血常规须具备以下三项中的两项:中性粒细胞绝对值<0.5×10 9 /L;血小板数<20×10 9 /L;网织红细胞绝对值<20×10 9 /L。其中中性粒细胞<0.2×10 9 /L者称极重型再障(very severe aplastic anemia,VSAA)。我国早年以急性或慢性再障(chronic aplastic anemia,CAA)分型。1987年第四届全国再障学术会议上将急性再障称为重型再障Ⅰ型,慢性再障后期发生恶化者称为重型再障Ⅱ型。
(一)SAA
起病急,进展迅速,常以出血和感染、发热为首发及主要表现。病初贫血常不明显,但随着病程呈进行性进展。几乎均有出血倾向,60%以上有内脏出血,主要表现为消化道出血、血尿、眼底出血(常伴有视力障碍)和颅内出血。皮肤、黏膜出血广泛而严重,且不易控制。病程中几乎均有发热,系感染所致,常在口咽部和肛门周围发生坏死性溃疡,从而导致败血症。肺炎也很常见。感染和出血互为因果,使病情日益恶化,如仅采用一般性治疗多数在1年内死亡。
(二)CAA
起病缓慢,以贫血为首发和主要表现;出血多限于皮肤黏膜,且不严重;可并发感染,但常以呼吸道为主,容易控制。若治疗得当、坚持不懈,不少患者可获得长期缓解甚至痊愈,但也有部分患者迁延多年不愈,甚至病程长达数十年,少数到后期出现SAA的临床表现。
【辅助检查】
(一)血常规
呈全血细胞减少,贫血属正常细胞型,亦可呈轻度大红细胞型。外周血片手工分类十分重要,红细胞形态应基本正常,仅见轻度大小不一,但无明显畸形及多染现象,无幼红幼粒细胞出现。网织红细胞显著减少。
(二)骨髓象
应做多部位骨髓穿刺涂片检查并同时进行骨髓小粒分类计数。SAA呈多部位增生减低或重度减低,三系造血细胞明显减少,尤其是巨核细胞和幼红细胞;非造血细胞增多,尤为淋巴细胞增多。CAA不同部位穿刺所得的骨髓象很不一致,可从增生不良到增生象,但至少要有一个部位增生不良;如增生良好,晚幼红细胞(炭核)比例常增多,其核为不规则分叶状,呈现脱核障碍,但巨核细胞明显减少。CAA可有轻度红系病态造血,但绝对不会出现粒系和巨核细胞病态造血。骨髓涂片肉眼观察油滴增多,骨髓小粒镜检非造血细胞和脂肪细胞增多,一般在60%以上。
(三)骨髓活组织检查和放射性核素骨髓扫描
由于骨髓涂片易受周围血液稀释的影响,有时一两次涂片检查难以正确反映造血情况,而骨髓活组织检查(至少取2cm骨髓组织)估计增生情况优于涂片,可提高诊断的正确性,应作为诊断再障必备条件。
再障骨髓病变的特点是造血组织减少,造血组织与脂肪组织比例多在2∶3以上。造血灶中造血细胞(指粒、红和巨核细胞系统)减少,而“非造血细胞”(指淋巴、浆、组织嗜碱和网状细胞)增多。骨髓中有血浆渗出、出血及间质水肿。SAA骨髓病变发展迅速而广泛;CAA则呈渐进性“向心性萎缩”,先累及髂骨,然后是棘突与胸骨。CAA尚存在代偿性增生灶,后者主要是幼红细胞增生伴成熟障碍。硫化 99 m 锝或氯化 111 铟全身骨髓γ照相可反映功能性骨髓的分布,可以间接反映造血组织减少的程度和部位。
(四)其他检查
流式细胞术检测骨髓CD34 + 细胞数对鉴别再障和骨髓增生异常综合征(myelodysplastic syndrome,MDS)有重要意义,再障显著降低(<0.5%),低增生MDS则明显增高。造血祖细胞培养不仅有助于诊断,而且有助于检出有无抑制性淋巴细胞或血清中有无抑制因子。成熟中性粒细胞碱性磷酸酶活力增高,血清溶菌酶活力减低。抗碱血红蛋白量增多。染色体检查除Fanconi贫血染色体畸变较多外,一般再障属正常,如有核型异常须除外MDS。
【诊断和鉴别诊断】
1987年第四届全国再障学术会议修订了再障诊断标准并于2007年再次修订如下:①全血细胞减少,网织红细胞绝对值减少,淋巴细胞相对增多。②骨髓检查显示至少有一个部位增生减低或重度减低(如增生活跃,巨核细胞应明显减少及淋巴细胞相对增多,骨髓小粒成分中应见非造血细胞增多)。有条件者应做骨髓活检(显示造血组织减少,脂肪组织增加)。③能除外其他引起全血细胞减少的疾病,如阵发性睡眠性血红蛋白尿、骨髓增生异常综合征、自身抗体介导的全血细胞减少、急性造血功能停滞、意义未定特发性血细胞减少(idiopathic cytopenia of uncertain significance,ICUS)、骨髓纤维化、急性白血病、恶性组织细胞病等。
再障必须和下列疾病相鉴别:
(一)阵发性睡眠性血红蛋白尿(PNH)
尤其是血红蛋白尿不发作者极易误诊为再障。本病出血和感染较少见,网织红细胞增高,骨髓幼红细胞增生,尿中含铁血黄素、糖水试验、Ham试验及蛇毒因子溶血试验呈阳性反应,成熟中性粒细胞碱性磷酸酶活力低于正常,外周血红细胞、中性粒细胞或淋巴细胞CD59和CD55标记率测定至少有二系血细胞CD59/CD55缺失率>10%及Flaer检测等,均有助于鉴别。
(二)骨髓增生异常综合征(MDS)
其中难治性贫血型极易和不典型再障相混淆,尤其是低增生MDS(骨髓活检造血细胞面积60岁以下<30%,60岁以上<20%)。MDS虽有全血细胞减少,但骨髓三系细胞均增生,巨核细胞也增多,三系均可见病态造血,染色体检查核型异常占31.2%,骨髓组织切片检查可见“幼稚前体细胞异常定位”(ALIP)现象。低增生MDS骨髓增生减低,染色体检查出现-5/5q - 、-7/7q - 、inv(3)等典型 MDS异常核型,但原始细胞数已>1%~20%,再障不应发现原始细胞。
(三)低增生性急性白血病
多见于老年人,病程缓慢或急进,肝、脾、淋巴结一般不肿大,外周呈全血细胞减少,未见或偶见少量原始细胞。骨髓灶性增生减低,但原始细胞百分比已达白血病诊断标准。
(四)纯红细胞再生障碍性贫血
溶血性贫血的再障危象和急性造血停滞,可呈全血细胞减少,起病急,有明确诱因,去除后可自行缓解,后者骨髓象中可出现巨原红细胞。慢性获得性纯红再障如有白细胞和血小板轻度减少,需注意和CAA作鉴别。
【治疗】
包括病因治疗、支持疗法和促进骨髓造血功能恢复的各种措施。以自身免疫性再障为例,非重型如不必依赖输血者治疗可以雄激素为主,辅以其他综合治疗,不少病例血红蛋白恢复正常,但血小板长期处于较低水平,临床无出血表现。输血依赖的非重型再障首选环孢素(Cs A)+雄激素治疗,6个月治疗无效者亦可选用ATG/ALG+CsA治疗。SAA预后差,一旦确诊宜及早(3周内)选用骨髓移植或ATG/ALG+CsA治疗。治疗方案的选择可依据患者年龄及有无HLA相配同胞供者(表16-2-3)。
(一)免疫抑制剂治疗(immunosuppresive therapy,IST) 适用于年龄大于50岁或无HLA相配同胞供髓者的SAA。>60岁患者慎用ATG。最常用的是抗胸腺球蛋白(ATG)和抗淋巴细胞球蛋白(ALG)。其机制主要通过去除抑制性T淋巴细胞对骨髓造血的抑制,其对B细胞无作用。剂量因来源不同而异,马源ALG/ATG 10~15mg/(kg·d),兔源 ALG/ATG 2.5~3.75mg/(kg·d),猪源ATG 20~30mg/(kg·d),共5天;用生理盐水稀释后先做过敏试验(单支ATG/ALG的1/10量加入生理盐水100ml静脉滴注1小时),如无反应,缓慢从大静脉内滴注,每天分2次,每次6~8小时;同时静脉滴注氢化可的松4mg/(kg·d),经另一静脉通道与ATG/ALG同步输注。患者应给予保护性隔离。为预防血清病,宜在第5天后口服泼尼松1mg/(kg·d),第15天后每5天减半,第30天停用。起效时间一般在用药后6~9个月,无效确认后可进行第2次ALG/ATG治疗,须换用其他动物来源的制剂。单用治疗SAA的有效率可达40%~60%,有效者50%可获长期生存。不良反应有发热、寒战、皮疹等过敏反应,以及中性粒细胞和血小板减少引起的感染和出血,滴注静脉可发生静脉炎,血清病在治疗后7~10天出现。用药期间维持血小板>10×10 9 /L。因ALG/ATG具有抗血小板活性作用,故不能在输注ALG/ATG的同时输注血小板悬液。
表16-2-3 获得性原发性SAA治疗方案的选择 *

注: * IST(immunosuppresive therapy):免疫抑制治疗;MSBMT(matched sibling bone marrow transplant):HLA相配同胞供者骨髓移植;MUDT(matched unrelated donor transplant):HLA相配非血缘供者移植;CBT(umbilical cord blood transplantation):脐血移植
环孢素(Cs A)的作用机制主要通过阻断IL-2受体表达来阻止细胞毒性T淋巴细胞的激活和增殖,抑制产生IL-2和γ干扰素。剂量为3~5mg/(kg·d),分两次口服。多数病例需要长期维持治疗,减量要缓慢,减量过快会增加复发风险。一般推荐疗效达平台期后持续服药至少12个月,以后逐渐减量,总疗程2~3年。对SAA的有效率也可达40%~60%,出现疗效的时间至少要3个月。不良反应有消化道症状、肝肾毒性作用、多毛、牙龈肿胀、肌肉震颤,因安全血药浓度范围较窄宜采用血药浓度监测,过去常采用测定全血CsA谷浓度(C0)来指导用药,安全有效谷浓度范围成人为150~250μg/L,儿童为100~150μg/L,近年多采用CsA的峰值(用药后2小时的血浓度C2),C2要比C0高5~10倍。
现代强烈免疫抑制治疗(指ALG/ATG和CsA联合治疗,CsA口服可与ALG/ATG同时应用或ALG/ATG开始后4周用)已成为SAA的标准治疗,有效率可达70%~80%,并且有效速度略快于单用ATG,强烈免疫抑制治疗的疗效已可和骨髓移植相近,但前者不能根治,且有远期并发症,如出现克隆性疾病,包括MDS、PNH和白血病等。伴有明显PNH克隆(>50%)的再障患者慎用ALG/ATG治疗;妊娠期不推荐使用ALG/ATG,但可予CsA治疗;先天性再障对IST无效。
其他免疫抑制剂尚有单克隆抗T细胞抗体(如抗CD52单克隆抗体,alemtuzumab)及吗替麦考酚酯(麦考酚酸酯,骁悉)等。大剂量静脉输注免疫球蛋白(HD-IVIg),可封闭单核-巨噬细胞Fc受体,延长抗体包裹血小板的寿命,亦可封闭抑制性T淋巴细胞的作用,中和病毒和免疫调节效应,适用于SAA有致命出血表现伴血小板同种抗体阳性、血小板输注无效时,以及病毒相关性严重再障的治疗。国外有应用大剂量环磷酰胺[CTX 45mg/(kg·d),连续4天]治疗SAA,但治疗相关病死率高而未被推荐。但上述免疫抑制剂的疗效均不及ALG/ATG和CsA。
(二) 造血干细胞移植(hematopoietic stem cell transplantation,HSCT)是治疗SAA和VSAA的最佳方法,且能达到根治目的。移植后长期无病存活率可达60%~80%,但移植需尽早进行,因初诊者常输红细胞和血小板,这样易使受者对献血员的次要组织相容性抗原致敏,导致移植排斥的发生率升高。一旦确诊SAA或VSAA,具有HLA配型相合的同胞供者,年龄<35岁,应首选同胞供者造血干细胞移植(MCD-HSCT);年龄在35~50岁的患者,应于2个疗程标准免疫抑制剂治疗失败后才考虑移植治疗。HLA配型相合无关供者的HSCT适应证掌握必须严格,仅适用于无同胞供者,且免疫抑制治疗失败患者的二线治疗。近年来,国内临床研究发现随着HLA配型技术的发展,预处理方案的改进及移植后支持疗法的加强,亲缘半相合造血干细胞移植(Haplo-HSCT)、无关供者 HSCT(UD-HSCT)和脐血 HSCT(UCB-HSCT)疗效与 MSDHSCT无明显差异,UCB-HSCT虽然造血重建率明显低于其他移植方式,但总体预后亦无明显差异。
(三)雄激素 系治疗CAA不必依赖输血患者和先天性再障的首选药物。常用的雄激素有司坦唑醇(康力龙,stanozolol),系17α烷基雄激素类;丙酸睾酮和十一酸睾酮(安雄)系睾丸素酯类。两者对造血干细胞具有直接刺激作用,促使其增殖和分化。
因此雄激素必须在一定量残存的造血干细胞基础上才能发挥作用,SAA常无效。但有端粒缩短的再障患者有效。丙酸睾酮50~100mg/d肌内注射,司坦唑醇6~12mg/d口服,安雄120~160mg/d口服,十一酸睾酮注射液0.25g肌内注射,每周1次,首次1.0g。疗程至少6个月以上。红系疗效较好,一般治疗后1个月网织红细胞开始上升,随后血红蛋白上升,2个月后白细胞开始上升,但血小板多难以恢复。部分患者对雄激素有依赖性,停药后复发率达25%~50%,复发后再用药仍可有效。丙酸睾酮的男性化副作用较大,肌注多次后局部常发生硬块,宜多处轮换注射。17α-烷基类雄激素的男性化副作用较丙睾为轻,但肝毒性反应显著大于丙睾,多数患者服药后出现谷丙转氨酶升高,严重者发生肝内胆汁淤积性黄疸,少数甚至出现肝血管肉瘤和肝癌,但停药后可消散。
(四)其他治疗 凡有可能引起骨髓损害的物质均应设法去除,禁用一切对骨髓有抑制作用的药物。积极做好个人卫生和护理工作。对粒细胞缺乏者宜保护性隔离,积极预防感染。输血要掌握指征,准备做骨髓移植者,移植前输血会直接影响其成功率,尤其不能输家族成员的血。一般以输入浓缩红细胞为妥。严重出血者宜输入浓缩血小板,采用单产或HLA相合的血小板输注可提高疗效。拟行异基因造血干细胞移植者应输注辐照或过滤后的红细胞和血小板悬液。反复输血有铁过载者宜应用去铁胺治疗。重组人EPO无益于再障的治疗,G-CSF可用于再障粒细胞缺乏的治疗,TPO和TPO受体激动剂对升高血小板也有一定疗效,TPO受体激动剂艾曲波帕还能用于难治性再障改善造血,联合IST治疗可提高疗效。中医药:治宜补肾为本,兼益气活血。常用中药为鹿角胶、仙茅、仙灵脾、黄芪、生熟地、首乌、当归、苁蓉、巴戟、补骨脂、菟丝子、枸杞子、阿胶等。

(资源61) “疑难血液病临床和细胞形态学讨论”之一(病例)
二、纯红细胞再生障碍性贫血
纯红细胞再生障碍性贫血(pure red cell aplasia,PRCA)简称纯红再障,系选择性影响骨髓红系前体细胞增殖和分化,引起单纯红系造血衰竭,而髓系、巨核系、淋系正常。单系造血衰竭综合征尚见于粒细胞系和巨核细胞系,前者即再生障碍型粒细胞缺乏症(见本篇第六章第一节“中性粒细胞减少与粒细胞缺乏症”),后者即获得性无巨核细胞性血小板减少性紫癜(acquired amegakaryocytic thrombocytopenic purpura),临床上以纯红再障最常见。
获得性纯红再障共同的临床表现是有严重进行性贫血,呈正常红细胞性或轻度大红细胞性贫血,伴网织红细胞显著减少(<1%,绝对值<10×10 9 /L),白细胞和血小板数正常或接近正常;骨髓有核细胞并不减少,粒细胞和巨核细胞系列增生正常,但幼红细胞系列显著减少,甚至完全缺乏,应<5%。个别病例可见幼红细胞系列成熟停顿于早期阶段,出现原红细胞小簇且伴巨幼样变,但缺乏较成熟的幼红细胞。临床有两种类型:
(一)急性自限型获得性纯红再障
多数以急性造血功能暂时停顿为主要表现。起病急,常在轻度感染后突然发生进行性全血细胞减少,网织红细胞减少,其血常规和骨髓象和再生障碍性贫血不能区别,有时在骨髓中出现巨大原始红细胞,经2~6周后自然恢复,恢复期网织红细胞上升,甚至出现反跳,骨髓象逐渐出现各期幼红细胞,称为急性造血停滞(acute arrest of hemopoiesis),如在原有慢性溶血性贫血基础上发生,原有溶血性黄疸亦可减轻,又称为溶血性贫血的再生障碍危象(aplastic crisis)。其病因多数为病毒感染,特别是人类微小病毒(parvovirus)HPV-B19感染,可选择性感染和溶解红系祖细胞,溶血性贫血因骨髓红系增生,易为HPV-B19所侵犯,尤其是患者有免疫缺陷时,可测定血清HPV-B19的IgG、IgM抗体,两者均阳性表示有近期感染,最好测定病毒的DNA序列。再障危象亦可发生在EB病毒、肝炎病毒和腮腺炎病毒感染基础上。药物也能引起急性造血停滞,如磺胺类药物、氯霉素、苯妥英钠、异烟肼、硫唑嘌呤等,通过免疫机制或直接抑制造血细胞DNA合成引起。急性纯红再障尚可见于1~4岁小儿,数周后自愈,并无感染因素,称儿童暂时性幼红细胞减少症(transient erythroblastopenia of childhood,TEC)。病毒感染引起应选用大剂量静脉用丙种球蛋白[400mg/(kg·d),每3~4周1次]。药物引起应及时停药,停药后大多数病例会完全恢复。
(二))慢性获得性纯红再障
主要见于成人,多数系自身免疫机制导致PRCA,可通过T淋巴细胞或NK细胞介导,少数可通过自身抗体介导的红系抑制。可分原发性PRCA(原因不明,约占50%的病例)和继发性PRCA。后者可与许多疾病相关,包括:①10%~15%的患者伴有胸腺瘤(thymoma),仅5%的胸腺瘤患者有纯红再障;这些胸腺瘤多数系良性,少数为恶性;女性多见(女∶男为3.0~4.5∶1)。因此慢性型PRCA均应详细检查有无胸腺瘤,必须进行CT扫描,胸腺瘤诊断一旦确立应尽早切除,术后贫血的缓解率仅30%;如术后未获缓解者,应给予免疫抑制剂治疗。②淋巴细胞增殖性疾病:PRCA见于6%的慢性淋巴细胞白血病,10%~15%的T细胞大颗粒淋巴细胞白血病,偶见于非霍奇金淋巴瘤和急性淋巴细胞白血病。③结缔组织病:PRCA可见于系统性红斑狼疮、类风湿关节炎、干燥综合征、混合结缔组织病、成人Still病。④重组人红细胞生成素诱发的纯红再障(epo-PRCA):主要见于长期应用基因重组人红细胞生成素(epoietin-alpha)皮下注射后发生PRCA,体内产生对PRCA的中和抗体。诊断epo-PRCA应至少使用EPO三周以上,血清检出EPO抗体,并有中和EPO的能力,疑epo-PRCA应及时停药。⑤骨髓增生异常综合征(MDS):PRCA亦可是 MDS首发表现,此系红系生成异常的克隆性疾病,并非免疫机制引起。
本病的治疗应及时选用免疫抑制剂。自身免疫因素引起的PRCA,包括慢性淋巴细胞白血病或自身免疫性疾病,发现有抑制红系和中和EPO的抗体,可选用肾上腺皮质激素治疗。若为恶性肿瘤伴发者应积极治疗原发病。对T细胞介导的慢性获得性纯红再障应选用环孢素、抗淋巴细胞球蛋白或抗胸腺细胞球蛋白、环磷酰胺等免疫抑制剂,雷公藤总苷也可选用,环孢素的疗效高于再生障碍性贫血,应作为首选。应用免疫抑制剂治疗可使66%以上的患者获得缓解,但复发率可达80%。体内抗体滴度高者也可选用血浆置换术。达那唑(炔羟雄烯异
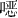 唑,danazol)或 CD20单克隆抗体(美罗华)亦可试用。为改善症状可输红细胞,长期反复输血铁过载者,宜及时选用去铁胺。
唑,danazol)或 CD20单克隆抗体(美罗华)亦可试用。为改善症状可输红细胞,长期反复输血铁过载者,宜及时选用去铁胺。
三、遗传性骨髓衰竭综合征
遗传性骨髓衰竭综合征包括Fanconi贫血(Fanconi anemia,FA)先天性角化不良(dyskeratosis conigenita,DC),Diamond-Blackfan贫血(Diamond-Blackfan anemia,DBA)、Shwachman-Diamond 综合征(Shwachman-Diamond Syndrome,SDS)等,其共同特点:①虽见于儿童期,但均可在成年期被诊断;②均有染色体或基因检查的异常;③部分病例可合并先天性畸形;④均容易发生恶性肿瘤,包括 MDS/AML;⑤治疗方针不同于获得性骨髓衰竭症,采用免疫抑制剂治疗无效,如用常规造血干细胞移植治疗反而引起高病死率,特别是FA和DC。
最常见为FA,多数系常染色体隐性遗传性疾病,有家族性。贫血多发现于5~10岁,多数病例伴有先天性畸形,特别是骨骼系统,如拇指短小或阙如、多指、桡骨缩短、体格矮小、小头、眼裂小、斜视、耳聋、肾畸形及心血管畸形等,皮肤色素沉着也很常见。本病血红蛋白F(HbF)常增高。染色体异常发生率高,可见染色体断裂、缺失、染色单体互换、核内再复制、环形染色体畸变等。淋巴细胞培养加入DNA交联剂可显示大量染色体断裂,借此确诊患者中,37%无先天性畸形,31%无贫血,7%两者皆无,故可诊断出不典型病例。患者DNA修复机制有缺陷,发现至少有16种不同的DNA修复基因(FANCA,FANCB,FANCC等)与本病有关,因此恶性肿瘤特别是白血病的发生率显著增高。10%的患儿双亲有近亲婚配史。可借助皮肤成纤维细胞或外周血淋巴细胞作染色体断裂试验诊断。
DC呈常染色体隐性遗传,多为10岁以下儿童发病,除全血细胞减少外,常具有指甲营养不良、皮肤色素沉着、口腔黏膜白斑三联征;已分离鉴定10个基因与DC发病有关,包括 DKC1 , TERC , TERT , TINF2 等,这些基因编码的蛋白和维持染色体端粒有关,因此其端粒长度变短,可借助淋巴细胞端粒长度测定诊断。SDS呈常染色体隐性遗传,80%病例有 SBDS 基因突变,临床特征为胰腺外分泌功能不全和骨髓衰竭。
上述三类遗传性骨髓衰竭综合征均呈再生障碍性贫血表现,而DBA呈现纯红再障表现。DBA90%于出生到1岁内起病。10%~20%的患者有家族史,为常染色体显性遗传,少数为隐性遗传。累及至少有 RPS17 , RPS19 , RPL5 , RPL11 等10个基因,这些基因编码核糖体蛋白。患儿生长发育迟缓,可有先天性畸形,如骨骼,心血管或泌尿生殖器畸形。患者红细胞腺苷脱氨酶(ADA)和HbF升高。80%的患儿对小剂量肾上腺皮质激素[泼尼松2mg/(kg·d),口服]有效,白介素-3[5~10μg/(kg·d),皮下注射]或甲氧普胺(10mg,每日3次口服,共4个月)亦有效。无效者可做HLA相合同胞供者异基因骨髓移植。
主要参考文献
1.卢静,吴德沛,胡绍燕,等.不同方式异基因造血干细胞移植治疗63例重型再生障碍性贫血患者的预后比较.中华血液学杂志,2015,36(8):633-636.
2.施均,郑以州.再生障碍性贫血与低增生性骨髓增生异常综合征鉴别诊断思路.中华血液学杂志,2013,34(10):910-912.
3.Goldman L,Schafer AI.Goldman-Cecil Medincine.25th ed.Philadelphia:Elsevier Saunders,2016,1114-1121.
4.Wilson DB,Link DC,Mason PJ,et al.Inherited bone mar-row failure syndromes in adolescents and young adults.Ann Med,2014,46(6):353-363.
第三节
阵发性睡眠性血红蛋白尿
王小钦
阵发性睡眠性血红蛋白尿(paroxysmal nocturnal hemoglobinuria,PNH)系获得性造血干细胞基因突变引起血细胞膜缺陷所致的慢性血管内溶血,常在睡眠时加重,可伴发作性血红蛋白尿、潜在的骨髓衰竭和血栓形成。
【病因与发病机制】
PNH是一种获得性造血干细胞克隆性疾病。位于X染色体上的 PIG-A 基因发生突变,使造血干细胞及其分化成熟的各种血细胞生成糖化磷脂酰肌醇锚(GPI-Anchor)障碍,使得需要借助于GPI锚才能连接在细胞膜上的一组膜蛋白称为GPI锚连蛋白(GPI-AP)缺失(图16-2-4),其中包括一些补体调节蛋白,使异常血细胞对补体敏感而破溶。正常红细胞有补体调节蛋白,能够保护红细胞免受自身补体的攻击,而PNH红细胞表面缺乏18种以上补体调节蛋白,故易受活化补体的攻击发生溶血,其中研究较清楚的补体调节蛋白有两种:CD55和CD59。PNH引起溶血的主要原因是CD59缺乏,而CD55的功能主要是防止补体的继续激活和放大。

图16-2-4 糖化磷脂酰肌醇锚(GPI锚)结构的模式图
根据红细胞对补体敏感性的不同,将PNH红细胞分为三型:Ⅰ型(补体敏感度正常)、Ⅱ型(中度敏感)、Ⅲ型(高度敏感),溶血程度与对补体敏感的红细胞所占比例密切相关。若Ⅲ型细胞占红细胞总量的半数以上,则经常有血红蛋白尿;若主要为Ⅱ型细胞,则可见间断的血红蛋白尿。Ⅲ型红细胞CD55、CD59完全缺失,Ⅱ型部分缺失,Ⅰ型基本正常。
【临床表现】
本病虽少见,但近年发病有增多趋势。我国北方多于南方,半数以上发生于20~40岁的青壮年,个别发生于10岁以下及70岁以上(2~83岁)。男性患病多于女性。我国患者的临床表现与欧美病例有所不同,起病多隐匿缓慢,以贫血为首发症状较多,以血红蛋白尿起病者较少,血栓形成也比国外少。
(一)血红蛋白尿(hemoglobinuria)
约3/4患者在病程中可有血红蛋白尿发作,但以血红蛋白尿发作为首发症状仅占15.9%。可频发,也可偶发(发作间隔>2个月),有的病例仅有尿隐血偶然阳性。以贫血为首发表现者,多在发病后半年至2年后进入血红蛋白尿发作期,最初较轻,发作次数少,以后逐渐加重,发作频繁。血红蛋白尿一般在晨起较重,呈红葡萄酒或红茶色、酱油样,轻者可无任何不适,重者有气短、面色苍白,腰腹部疼痛和发热等症状。睡眠后晨起较重。血红蛋白尿的诱发因素有药物(铁、氯化铵、阿司匹林、呋喃妥因、氯丙嗪、苯巴比妥、磺胺药、青霉素、有机碘造影剂等)、病毒感染、输血、过度疲劳、情绪波动、大量饮酒、月经或妊娠期、疫苗接种、手术等。一般血浆游离血红蛋白浓度超过300~400mg/L时,出现血红蛋白尿,因慢性血管内溶血,含铁血黄素尿阳性。血红蛋白尿和持续的含铁血黄素尿导致不同程度的铁丢失。
(二)贫血
初诊时95%有贫血,54%呈全血细胞减少。PNH和再障关系相当密切,约25%~31%患者可以再生障碍性贫血起病,经过一定阶段出现PNH的表现;或患者以典型的PNH起病,以后在疾病过程中发生骨髓再生障碍;亦可PNH伴再障特征或再障伴PNH特征;都可称再障-PNH综合征,约占PNH病例的10%。
(三)感染
近半数患者易有继发感染,以支气管、肺部及泌尿生殖道感染较为常见,感染的原因与中性粒细胞减少及吞噬功能降低以及溶血导致单核-巨噬细胞系统封闭有关。感染可诱发溶血或引起再障危象。
(四)血栓形成
凡有严重溶血,具有较大PNH克隆的病例,易有血栓形成。欧美报道23.1%~49.7%患者在病程中可发生一次或一次以上血栓形成,而国内发生率(5.3%)较低,且血栓形成发生较晚,多为单发,主要在肢体表浅静脉,很少累及内脏血管,静脉比动脉多见,病情较轻,很少引起死亡。如病程中反复发生腹痛,可能与血栓形成有关,可见肠系膜血栓形成、脾栓塞、肝静脉血栓形成所致Budd-Chiari综合征;肺微血管血栓形成可致肺动脉高压;大脑静脉最易累及矢状窦血栓形成,有头痛、眼痛、眼乳头水肿、偏瘫等,个别见附睾静脉血栓形成、周围肢体静脉血栓形成、肾静脉血栓形成等。血栓形成的原因与多种危险因素有关,包括血小板缺失CD59、补体激活血小板功能异常,血管内反复溶血、一氧化氮(NO)的耗竭均可使血小板聚集增强,血浆凝血因子活性增高及纤溶受损等。
(五)肾脏损害
多数患者有不同程度血尿、蛋白尿及肾功能减退,多在起病5年内发生。X线检查见肾脏外形单侧或双侧增大,密度较高,皮质梗死、增厚,肾乳头坏死。在严重血红蛋白尿发作期,可发生急性肾衰竭。
(六)其他
因溶血产生大量游离血红蛋白使NO耗竭致平滑肌功能障碍,引起吞咽困难、食管痉挛、腹痛及勃起障碍。脾常中度增大,肝轻度或中度肿大,少数病例由于长期大量溶血形成胆色素性结石。也有报告本病可合并肿瘤,包括淋巴瘤和白血病。伴有妊娠常可致流产、死胎,且妊娠可以诱发溶血发作,增加血栓形成的发生率,对母体有生命危险。
【实验室检查】
(一)血常规
贫血程度轻重不一,重者血红蛋白低于50g/L,红细胞形态无特殊,伴溶血者可轻度大红细胞增多,大小不一,若伴缺铁,可见小细胞低色素改变。网织红细胞轻至中度增多,但不及其他类型溶血性贫血为高。中性粒细胞数常减少,感染时可升高,此与再障不同。白细胞碱性磷酸酶活力降低。血小板中至重度减少,约18%患者以出血为首发症状,约20%患者血小板数正常,血小板寿命多为正常。半数以上有全血细胞减少,以血红蛋白尿不发作组为著。
(二)骨髓象
呈增生象,红系增生;但也可增生在正常范围,甚至增生低下。骨髓象可存在二系或三系“病态造血”,故易误诊为骨髓增生异常综合征。
(三)尿
可有血红蛋白尿或尿潜血阳性,镜检无红细胞。含铁血黄素尿(Rous试验)常持续阳性。
(四)红细胞补体溶血试验
1.酸溶血试验(Ham test)
其原理是PNH红细胞在酸化血清(pH 6.4)条件下易被替代途径激活的补体破溶,正常红细胞则否。如加入镁(一般加用250mmol/L氯化镁10μl)(即酸溶血改良试验),可提高试验的灵敏度。本试验特异性高,但灵敏度比糖水试验差,约10.4%PNH患者酸溶血试验始终阴性,个别患者常反复多次检查,才呈阳性反应。如使用血型不合的血清或酸化过度,或遗传性红细胞生成异常性贫血Ⅱ型时,可出现假阳性,宜加鉴别。
2.糖水试验
本试验灵敏度高,特异性不及酸溶血试验。在PNH患者,溶血度一般为10%~80%,个别低达5%,某些白血病及骨髓纤维化患者,也可发生溶血,然溶血度一般<10%,故溶血度须超过30%才有诊断价值,在5%~10%为可疑。对酸溶血试验阳性的遗传性红细胞生成异常性贫血患者,糖水溶血试验则呈阴性。
3.蛇毒因子溶血试验
其原理是蛇毒因子能通过替代途径激活补体,对补体敏感的PNH红细胞发生溶血。本试验溶血度在一定程度上能反映PNHⅢ型红细胞的多少,与临床上溶血程度成平行关系。其特异性强,敏感性优于酸溶血试验,但低于糖水试验。
4.补体溶血敏感试验
检测使红细胞破溶所需的补体量,据此可将PNH细胞分为Ⅰ、Ⅱ、Ⅲ型,临床溶血轻重取决于Ⅲ型细胞的多少。诊断价值高,但方法繁琐。
(五)流式细胞术测定GPI锚连接蛋白
应用针对GPI锚连接蛋白包括CD55、CD59、CD16、CD67、CD24等的相应抗体做免疫荧光染色,以流式细胞仪检测并计数缺乏这类膜蛋白的异常细胞比例,是诊断PNH灵敏而特异的方法。PNH患者不论外周血红细胞、中性粒细胞或骨髓单个核细胞CD59阴性细胞均>10%,当CD55或CD59细胞占3%~5%时即可检出,远较酸溶血试验敏感。PNH克隆累及造血细胞次序为粒细胞→单核细胞→红细胞→淋巴细胞,骨髓PNH克隆出现比外周血早,建立PNH诊断至少有一系及以上细胞的两种GPI锚连蛋白缺失。CD59敏感度要高于CD55,CD59 - 粒细胞可最早被检出,有早期诊断价值,且不受输血影响。
(六)流式细胞术检测气单胞菌溶素前体变异体(Flaer)
Flaer是Alexa-488标记的无活性气单胞菌溶素前体的变异体,它同野生型前气单胞菌溶素相似,可特异地结合于GPI锚连蛋白,但并不形成细胞通道,不引起细胞溶血,因此不会导致细胞死亡。该标记类似于荧光素,可在一定条件下被激发出荧光,可以通过流式细胞仪进行检测,并区分GPI - 和GPI + 细胞。同传统的检测CD55、CD59相比,Flaer对检测微小PNH克隆非常敏感,且不受输血和溶血的影响,诊断PNH更敏感、更特异。
【诊断】
临床表现符合PNH;酸溶血、糖水、蛇毒因子或含铁血黄素尿试验中有任两项阳性,或仅一项阳性,但有2次以上阳性者;或流式细胞术发现外周血中CD55或CD59阴性中性粒细胞或红细胞>10%(5%~10%为可疑),即可诊断。
国际PNH工作组将PNH患者分为如下几类:①经典型PNH:该类患者有典型的溶血和血栓形成;②合并其他骨髓衰竭性疾病:如再生障碍性贫血(AA)或骨髓增生异常综合征(MDS);③亚临床型PNH:患者有微量PNH克隆,但没有溶血和血栓的实验室和临床证据。
本病漏诊、误诊率高,故须认真与遗传性球形红细胞增多症、自身免疫性溶血性贫血、葡萄糖-6-磷酸脱氢酶缺乏症所致的溶血、阵发性冷性血红蛋白尿、再生障碍性贫血、遗传性红细胞生成异常性贫血、MDS等相鉴别。
【治疗】
根治有赖于去除异常的克隆,适合者可作骨髓移植治疗,但不作为首选。治疗原则是促进正常造血功能的恢复,尽量避免诱发因素,控制急性溶血发作,防治并发症。
(一)骨髓移植
异基因骨髓移植是唯一可以治愈本病的方法,但PNH是种良性克隆性疾病,少数患者还可能自愈,而骨髓移植有一定风险,因此需谨慎考虑。目前认为仅适用于年轻、有重型再障或难以控制的重度溶血、反复血栓形成患者。移植成功率约50%~70%,但移植早期病死率甚高,可达33.3%。
(二)依库珠单抗(Eculizumab)
是一种人源化的单克隆抗体,可特异地与补体C5结合,阻止其活化,从而抑制膜攻击复合物的形成。对经典型PNH控制溶血,脱离输血依赖,防止血栓形成有明显疗效,已获得美国FDA和欧洲药物协会(EMEA)的批准应用于临床。具体用法:每周静脉输注600mg,共4周,然后900mg用1周,再每2周输注900mg,共6个月。但对再障-PNH综合征疗效不明显。要警惕脑膜炎球菌感染。
(三)免疫抑制剂单独或联合应用
ATG、ALG、环孢素等免疫抑制剂治疗对伴有骨髓增生不良的再障/PNH综合征有一定疗效,对经典型PNH无效。但必须注意ATG/ALG易诱发补体的激活。
(四)其他减轻溶血发作的方法
1.糖皮质激素 作用机制可能与抑制替代途径的补体激活有关。剂量为泼尼松20~60mg/d,溶血控制后可隔日口服15~40mg维持。急性溶血发作也可用氢化可的松100~200mg/d或地塞米松10~15mg/d静脉滴注,多数血红蛋白尿可在1~3天内消失,7天内尿潜血转阴,为防止复发,改口服泼尼松维持。约50%以上患者可能有效,若泼尼松应用6周后无效,可停用。
2.小剂量化疗 有应用6巯嘌呤或苯丁酸氮芥加泼尼松或长春新碱、环磷酰胺和泼尼松联合或美法仑加泼尼松治疗以抑制PNH干细胞,但多数报告认为疗效不确定。
3.维生素E 每日300mg,分3次口服,但效果并不肯定。
4.尽量减少血红蛋白尿的诱发因素,并口服NaHCO 3 3g/d。
(五)贫血的治疗
PNH贫血原因是多因素的,应视不同机制选择下列治疗:①PNH患者合并明确缺铁时,可以小剂量、短期补铁。口服铁剂一般为常规量的1/3,铁剂不会加剧溶血。②雄激素,促进红系造血。③溶血严重时需补充叶酸。④重度贫血时可以输洗涤红细胞。
(六)血管栓塞的治疗可用肝素抗凝治疗
但须注意小剂量肝素可激活补体,加重溶血,血栓形成恶化,因此须尽快地转为双香豆素类药物抗凝治疗。也有应用链激酶及尿激酶溶栓治疗PNH伴血栓形成。
(七)基因治疗
尚在研究和试验阶段。
【病程与预后】
本病多呈慢性过程,中数生存期约10年,也有长达20年以上,个别至43年。极少数可呈急性病程,发病后数月即死亡。其预后与补体敏感的红细胞量、骨髓再生障碍程度及有无并发症相关。国内主要死因是出血和感染。国外是血栓形成。某些PNH患者随着年龄增长,病情可减轻,甚至达到完全缓解。约5%患者最终可演变为急性粒细胞白血病,个别患者可演变为骨髓增生异常综合征。
主要参考文献
1.中华医学会血液学分会红细胞疾病(贫血)学组.阵发性睡眠性血红蛋白尿症诊断与治疗中国专家共识.中华血液学杂志,2013,34(3):276-279.
2.Brodsky RA.Paroxysmal nocturnal hemoglobinuria.Blood,2014,124(18):2804-2811.
第四节
缺铁性贫血和其他低色素性贫血
王小钦
一、缺铁性贫血
铁缺乏症(iron deficiency,ID)是体内长期铁负平衡的结果,最初引起体内贮存铁耗尽(iron depletion),继之红系细胞内发生缺铁,称为缺铁性红细胞生成(iron deficient erythropoiesis,IDE),最后才发生缺铁性贫血(iron deficiency anemia,IDA)。IDA是体内贮存铁缺乏影响血红素合成所引起的贫血,其特点是骨髓、肝、脾等器官组织中贮存铁减少,血清铁、运铁蛋白饱和度和血清铁蛋白降低,典型的呈小细胞低色素性贫血。它是一种综合征,并非一种疾病。
铁缺乏症是最常见的营养素缺乏症,至今仍是世界各国普遍而重要的健康问题,尤其是发展中国家,其高危人群为妇女、婴幼儿和儿童。据全球187个国家1990年至2010年间疾病负担数据证实铁缺乏影响了20亿人。铁缺乏症的患病率为IDA的2倍。据复旦大学各附属医院流行病学调查,ID的患病率:6个月~2岁婴幼儿达75.0%~82.5%,育龄妇女为43.32%,妊娠3个月以上妇女为66.27%,10~17岁青少年为13.17%;以上人群IDA的患病率分别为33.8%~45.7%、11.39%、19.28%及9.84%。
【铁代谢】
(一)铁稳态和铁分布
铁是人体最丰富的必需微量元素之一,所有具有功能的细胞均含有铁,它广泛参与机体内的代谢过程。缺铁可引起血红素合成障碍导致IDA,也会影响含铁酶包括线粒体中细胞色素酶系统的活力以及肌红蛋白的合成。同时,机体还必须防止游离铁的毒害作用,后者可促发产生大量自由基。因此,人体存在严格的铁代谢调节机制,可以确保体内铁始终处于正常生理水平,称机体的铁稳态(iron homeostasis)。人体铁代谢是在“封闭”系统内反复循环,铁重复被利用,衰老红细胞被巨噬细胞吞噬后,所释放的铁约80%以上被重新利用(图16-2-5)。除月经及上皮细胞脱落丢失铁外,人体无明显的生理性排泄铁,因此正常成年男性和绝经后妇女一般不会发生缺铁。
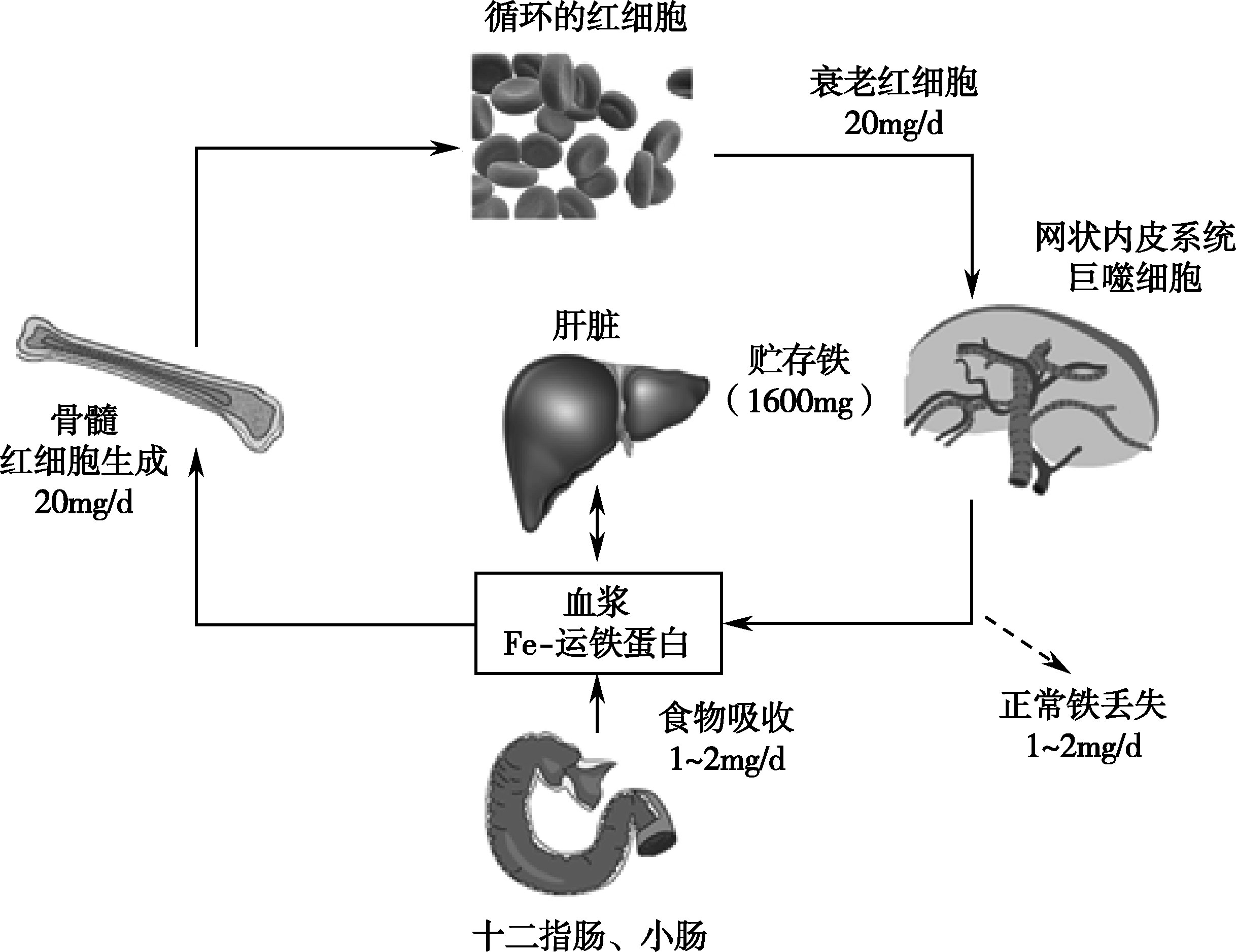
图16-2-5 正常人体铁稳态
正常成人含铁总量男性为50mg/kg,女性为40mg/kg。铁的62.1%组成血红蛋白,4%在肌红蛋白,31%以铁蛋白和含铁血黄素的形式储存,0.3%存在于参加细胞代谢的血红素酶类,0.1%为血液中的转运铁,2.5%为短暂结合于细胞膜或细胞间蛋白的易变池铁。成年女性的储存铁较男性显著减少,容易发生IDA。
(二)铁的吸收和转运
国人每日普通饮食中所供给的铁量为15~30mg,其中5%~10%被吸收,吸收量约1~2mg/d,主要吸收部位在十二指肠和空肠上段。铁的吸收形式有两种:①血红素铁,来自血红蛋白及动物食物的其他血红素蛋白,为二价铁,吸收率高。②非血红素铁,来自植物性食物中的高铁化合物等,为三价铁,必须转变为可溶性二价铁才易被吸收。胃酸可增加非血红素铁的溶解度,维生素C作为还原剂利于铁吸收。植物性食物中的磷酸盐、植酸盐、茶叶中的鞣酸及咖啡中的一些多酚类化合物等,都可与铁形成难以溶解的盐类而抑制非血红素铁的吸收,蛋黄中的磷蛋白和卵黄高磷蛋白和铁结合后可溶性差而不易吸收。因此铁的吸收率因食物种类而异,动物性食物约为20%~25%(蛋仅3%),植物性食物的吸收率<5%(但大豆为7%),人乳铁吸收率为50%,牛乳仅10%。
食物中三价铁还原为二价铁后,在二价金属离子转运体1(DMT1)介导下进入肠上皮隐窝细胞,穿过上皮细胞的基底膜进入血流,转变为三价铁后与运铁蛋白(ferroportin)结合,转运到肝和各组织。运铁蛋白主要由肝合成,分子量为79 500的糖蛋白,1分子运铁蛋白有2个结合三价铁的位点。正常人运铁蛋白血浆浓度为2.65~4.30g/L,应用间接法测定,即为总铁结合力(total iron binding capacity,TIBC),即血浆中能与铁结合的球蛋白的总量。正常情况下仅以其总量的1/3与铁结合,这部分称血清铁,2/3未与铁结合的运铁蛋白称为未饱和的运铁蛋白(图16-2-6)。运铁蛋白饱和度=血清铁/总铁结合力×100%。
图16-2-6 各种疾病血清铁和总铁结合力的比较
肝细胞膜上有两种运铁蛋白受体(transferrin receptor,Tf R):TfR1和TfR2。幼红细胞摄取运铁蛋白铁也需TfR,中幼红细胞TfR最多,受体可随细胞成熟而丢失。TfR是一种跨膜糖蛋白,铁被受体运送到幼红细胞内后,运铁蛋白和受体重新被运送到细胞表面,运铁蛋白被释放而重新被利用,铁被幼红细胞所摄取。进入幼红细胞的铁在线粒体上与原卟啉结合形成血红素,多余的铁以铁蛋白形式贮存于幼红细胞中。
铁的储存形式有铁蛋白和含铁血黄素两种。铁粒幼细胞中的铁颗粒即为聚合的铁蛋白。含铁血黄素是变性或部分去蛋白质的铁蛋白聚合形成的不溶性含铁复合物,骨髓中可染铁即分布于骨髓小粒的含铁血黄素。
(三)铁代谢的调节
铁调素(hepcidin)是调节体内铁稳态的重要的铁调节激素,是由肝脏产生的含25个氨基酸的多肽,可调节小肠铁吸收、巨噬细胞铁再循环及肝铁的动用。铁调素是一种小肠铁吸收和巨噬细胞、肝细胞铁释放的负调控因子。在高铁状态、感染、炎症时产生增加,缺铁、组织释放缺氧信号时产生减少。铁缺乏时,铁调素转录受抑,促进肠道铁吸收和体内贮存铁释放。贫血缺氧时,低氧诱导因子2α(HIF-2α)增加,肾脏产生促红细胞生成素(EPO)增多,从而刺激红系造血。HIF-2α增加了肠上皮细胞顶端DMT1的表达,以增加膳食铁从肠道吸收入肠上皮细胞。铁调素水平下降,运铁蛋白不再分解,肠上皮细胞基底膜铁转运增加,巨噬细胞贮存铁转运至血液循环增多。肝细胞对铁的释放较巨噬细胞为慢。总之,在缺铁状态下,铁调素通过增加肠道铁吸收、增加巨噬细胞贮存铁和肝铁的释放和转运来调节铁代谢。
十二指肠隐窝细胞可以感受体内铁变化,通过调节各种转运蛋白的表达量来控制铁的吸收。铁调节蛋白(IRP)可以通过调节TfR、运铁蛋白的合成水平来调节铁稳态。
【病因】
(一)营养因素
因饮食中缺乏足够量的铁或食物结构不合理,导致铁吸收和利用减低。国人膳食中供铁量并不少,但铁来源的食物构成不合理,仅20%的铁来源于动物食品。当生理性铁需要量增加时,如婴幼儿、青少年、妊娠妇女和哺乳期妇女,就容易发生营养性IDA。妇女一次月经平均失血量40~60ml,相当于20~30mg铁,因此需铁量比男性多,为2mg/d;妊娠期为供应胎儿所需及分娩时失血所丢失的铁,估计一次正常妊娠要额外增加960mg铁,妊娠中、后期需铁量达4~6mg/d,单纯从饮食中难以获得。
(二)慢性失血
慢性失血是IDA最常见的病因之一。如按每毫升血含铁0.5mg计算,慢性长期失血即使每天失血量少至3~4ml,也足以引起缺铁。IDA常是胃肠道肿瘤的首发表现,即使粪隐血试验阴性,也不能除外消化道出血,成年男性发生IDA一定要进行胃镜和肠镜检查。妇女缺铁的常见原因是月经量过多。在农村,钩虫感染是慢性消化道失血的原因。血尿、咯血、反复鼻衄、血红蛋白尿(如阵发性睡眠性血红蛋白尿、心脏人工瓣膜和行军性血红蛋白尿),也是慢性失血的原因。
(三)吸收障碍
常见于胃全切除和胃次全切除后数年发生缺铁。消化性溃疡长期服用H 2 受体拮抗药或质子泵抑制剂不致引起IDA,但萎缩性胃炎可影响铁的吸收。慢性腹泻、累及十二指肠和近端空肠的小肠疾病,不仅引起铁吸收不良,并且随着大量肠上皮细胞脱落而失铁。幽门螺杆菌(Hp)感染可能与宿主竞争可利用铁,减少铁的吸收。口服铁制剂常失效,Hp根治后,口服铁剂疗效恢复。
(四)遗传性
遗传性IDA甚为罕见,近年有一种常染色体隐性遗传的铁难治性IDA(iron-refractory iron deficiency anemia,IRIDA)被认识。由于TMPRSS6(一种Ⅱ型跨膜丝氨酸蛋白酶,可抑制激活hepcidin的信号通路)突变,导致Hepcidin高表达,阻断肠道铁吸收和铁再循环障碍,引起铁剂治疗无效的IDA。
【临床表现】
IDA的临床表现除因贫血引起组织器官缺氧导致贫血的一般性表现外,还有因组织缺铁导致的各种临床表现。因为许多影响细胞氧化还原过程的酶含有铁或为铁依赖酶,酶活力降低可产生多种临床表现:①可引起患儿精神发育和行为改变,这可能和单胺氧化酶活力降低、儿茶酚胺代谢紊乱有关;②劳动耐力降低,可能和细胞色素C及线粒体中α-甘油磷酸氧化酶活力降低、肌红蛋白量减少、影响骨骼肌氧代谢有关;③细胞免疫功能减弱,中性粒细胞杀菌能力减低;④抗寒能力降低,三碘甲腺原氨酸(T 3 )水平减低。严重IDA可致黏膜组织变化和外胚叶营养障碍,出现口炎、舌炎、萎缩性胃炎和胃酸缺乏,皮肤干燥、毛发干枯脱落、指甲扁平、脆薄易裂和反甲,甚至出现吞咽困难及异食癖。缺铁和感染的关系有待澄清,缺铁患儿易发生感染,但过量补铁后感染反而增多。
【实验室检查】
(一)血常规
铁缺乏症早期无贫血。IDA阶段贫血轻时呈正常红细胞性,严重时呈典型的低色素小细胞性贫血。成熟红细胞大小不一,中心淡染区扩大。红细胞分布宽度(RDW)>0.14。网织红细胞计数大多正常,亦可减低或轻度升高。白细胞计数正常,如近期内有大量出血,中性粒细胞和血小板数可增多。
(二)骨髓象
幼红细胞轻度或中度增生,中幼红细胞比例增多。贫血严重的患者幼红细胞体积偏小,核染色质致密,胞质减少、染色偏蓝,边缘不整齐,有血红蛋白形成不良的表现。骨髓铁染色显示骨髓小粒可染铁消失,铁粒幼红细胞低于15%。富含骨髓小粒的涂片铁染色缺乏可染铁,是诊断缺铁的金标准。
(三)血清铁和总铁结合力测定
在IDA时,血清铁<8.95μmol/L(50μg/dl),总铁结合力(TIBC)>64.44μmol/L(360μg/dl),运铁 蛋白饱和度(transferrin saturation,TS)<0.15。血清铁并非是缺铁的灵敏指标,且有昼夜变化,早晨高而夜间低,炎症性疾病、结缔组织病和恶性肿瘤都可使血清铁降低,肝细胞坏死可使血清铁升高。TIBC测定值较稳定。运铁蛋白饱和度<0.15是IDE的指标。
(四)血清和红细胞内碱性铁蛋白测定
常用放射免疫双抗体法测定,红细胞铁蛋白要先分离纯化红细胞制备悬液。血清铁蛋白(serum ferritin,SF)和体内贮铁相关性极好,1μg/L的SF相当于8~21mg贮铁,可作为贮铁缺乏的指标。诊断单纯性缺铁一般认为SF<20μg/L表示贮铁减少,<12μg/L为贮铁耗尽;红细胞碱性铁蛋白<6.5μg/细胞作为缺铁指标。SF系反映缺铁较敏感的指标,可用于早期诊断和人群的筛检,诊断IDA的敏感度为92%,特异度为83%。但SF易受感染、炎症、结缔组织病、肿瘤和肝疾病的影响而升高,而红细胞碱性铁蛋白则较少受上述因素的影响,更能正确地反映贮铁状态。
(五)红细胞游离原卟啉和血液锌原卟啉测定
缺铁时锌原卟啉(ZPP)和红细胞游离原卟啉(FEP)均可升高。FEP和ZPP升高尚见于铅中毒、慢性感染、炎症、恶性肿瘤和铁粒幼细胞性贫血等。
(六)血清运铁蛋白受体(sTfR)测定
是迄今反映IDE的最佳指标,sTfR水平不受炎症、肝病和妊娠等因素的影响,可以较正确地反映缺铁,因此可用于妊娠期缺铁和慢性病贫血合并缺铁的诊断,其灵敏度和特异度均优于SF。一般sTfR浓度>26.5 nmol/L(2.25μg/ml)可诊断缺铁。sTfR的水平也可反映贫血患者骨髓幼红细胞的生成情况。有认为采用复合参数如sTfR/SF和sTfR/logSF,尤其是后者更有助于慢性病贫血伴缺铁的诊断。
(七)网织红细胞血红蛋白量(reticulocyte hemoglobin content,CHr)测定
诊断缺铁的标准为<28pg。
【诊断与鉴别诊断】
1.IDA的诊断
IDA的诊断包括两个方面:确立是否系缺铁引起的贫血和明确引起缺铁的病因。典型的IDA诊断不难,可根据病史、典型的低色素性贫血形态学改变以及缺铁指标阳性而获得诊断。国内诊断标准如下:①小细胞低色素贫血,成年男性血红蛋白(Hb)<120g/L,女性Hb<110g/L,妊娠妇女<100g/L;平均红细胞容积<80fl,平均血红蛋白含量<27pg,平均血红蛋白浓度<0.32。②血清铁蛋白<12μg/L,血清铁<8.95μmol/L,运铁蛋白饱和度<0.15,总铁结合力>64.44μmol/L。③红细胞游离原卟啉>0.9μmol/L或血液锌原卟啉>0.96μmol/L,或红细胞游离原卟啉/血红蛋白>4.5μg/g Hb。④血清可溶性运铁蛋白受体(sTf R)>26.5nmol/L。⑤骨髓铁染色提示骨髓小粒可染铁消失,铁粒幼红细胞<15%。符合第1条和2~5条中任何一条,可诊断为IDA。
对早期缺铁的诊断需借助于实验室检查,常用的诊断标准为:单一SF≤12~20μg/L为贮铁缺乏,如加上下列三项指标[FEP>1.78μmol/L(100μg/dl)全血,FEP/Hb>4.5μg/gHb和TS<0.15]中两项异常,即可诊断为IDE。铁剂治疗试验也是确定本症的方法之一。
2.铁难治性IDA
遗传性铁难治性IDA的诊断主要根据以下几条:①小细胞低色素性贫血,贫血往往呈轻中度,故有时在成人期才被发现。红细胞数量会有轻度增多,网织红细胞降低。②铁代谢异常,血清铁和运铁蛋白饱和度下降,而多数患者的铁蛋白正常。③铁调素正常或升高。④口服铁剂无效,静脉补铁起效慢,贫血仅能部分纠正。⑤有家族遗传性,基因检测可发现 TMPRSS6 突变,存在多种基因突变类型,如 S304L 、 K225E 、 K253E 、 G228D 、 R446W 、 V736A 等。⑥排除营养性IDA及其他遗传性IDA,如无运铁蛋白血症、二价金属转运蛋白Ⅰ(DMTⅠ)突变等。
3.鉴别诊断
低色素性贫血(hypochromic anemia)可见于珠蛋白生成障碍性贫血、血红蛋白病和铁粒幼细胞性贫血等。功能性缺铁(functional iron deficiency)指患者体内总铁量并不少,但铁被锁定在巨噬细胞,不能释放供幼红细胞合成Hb用,常见于慢性病贫血和肾衰长期血透患者,也可有小细胞低色素贫血,都需注意鉴别。珠蛋白生成障碍性贫血和血红蛋白病,血清铁、TS、SF和骨髓可染铁均增多。铁粒幼细胞性贫血血清铁增高而TIBC降低,骨髓涂片铁染色可见典型的环形铁粒幼细胞。慢性病贫血(ACD)血清铁减低,TIBC正常或减低,SF正常或增高,骨髓小粒可染铁增多,铁粒幼细胞减少。MCV<72fl者ACD甚罕见,而IDA则常见。
【治疗】
(一)病因治疗
病因治疗相当重要,因为IDA是一种综合征,不能只顾补铁治疗而忽略其基础疾病的治疗,例如延误了胃肠道肿瘤的诊断和治疗,其后果不堪设想。
(二)口服铁剂
是治疗IDA的首选方法。口服铁剂的种类很多(表16-2-4),可分三类:无机铁、有机铁及血红素铁。至今仍认为硫酸亚铁是口服铁剂中的标准制剂,但它是无机铁剂,故胃肠反应大,主要和含有的游离铁离子有关。有机铁剂反应小,其中以多糖铁复合物最小;琥珀酸亚铁不仅含铁量高且吸收好,生物利用度高,不良反应又小,较常用。成人治疗剂量以每天150~200mg元素铁为宜,预防剂量每天10~20mg元素铁。为减少硫酸亚铁的胃部刺激反应,宜在餐后服用。较大剂量维生素C(每30mg铁剂至少口服200mg)或琥珀酸可增加铁剂的吸收,铁剂忌与茶同服,钙盐及镁盐亦可抑制铁吸收,应避免同时服用。
表16-2-4 常用口服铁剂
口服铁剂有效者网织红细胞在治疗后3~4天即开始上升,第10天达高峰,随后血红蛋白上升,一般需要治疗2个月左右,血红蛋白恢复正常。贫血纠正后至少需要继续治疗3个月或使SF恢复到50μg/L以补足贮存铁,总疗程一般需要3~6个月,否则易复发。
口服铁剂的不良反应有恶心、上腹痛、便秘和腹泻。如治疗3周无治疗反应,应检查诊断是否准确、是否按医嘱服药、有无活动性出血、有否铁吸收障碍、有否干扰铁吸收和利用的因素存在。
(三)注射铁剂
常用低分子右旋糖酐氢氧化铁复合物注射液、蔗糖铁注射液及葡萄糖酸铁钠注射液三种(表16-2-5)。注射铁剂推荐静脉注射。静脉注射过快(>100mg/min)可致局部静脉疼痛、发红及金属味,但时间很短,只要缓慢注射即可消失。全身反应包括即刻及延迟反应;即刻反应有低血压、头痛、恶心、荨麻疹,罕有过敏反应,但严重可致命;延迟反应包括淋巴结肿大、肌痛、关节痛、发热等。严重过敏反应甚少见,主要见于右旋糖酐铁,发生率约0.7%,葡萄糖酸铁钠为0.04%,蔗糖铁则更低。长期过量应用会增加氧化应激和感染的风险。
表16-2-5 三种注射铁剂的剂量
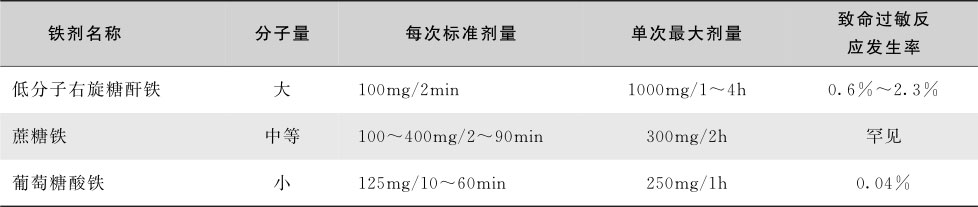
注射铁剂应严格掌握指征:①不能耐受口服铁剂或口服无效者,如胃大部切除、小肠旁路手术、乳糜泻、萎缩性胃炎、炎症性肠病等消化道疾病,遗传性铁难治性IDA;②需要尽快补铁患者,如妊娠后期的缺铁,失血速率快于口服铁剂的补偿率,先天性凝血出血疾病;③长期血透不能维铁平衡或有功能性缺铁患者(如慢性肾衰贫血)同时应用红细胞生成素治疗者。
注射铁剂总量可按下列公式计算:

静脉补铁后,4~5天网织红细胞数上升,1周后Hb上升,6周可达正常水平。如治疗目标为SF>50μg/L,常需4~6个月。
注射铁剂的具体用法如下:
低分子右旋糖酐氢氧化铁复合物注射液(Cosmofer,100mg/支),可以肌注、静脉推注和静脉滴注。静脉滴注首次使用前先做过敏试验,25mg溶于50ml生理盐水中,静脉滴注5分钟以上,如60分钟后无不良反应,即可静脉滴注。右旋糖酐铁100mg(2ml)用0.9%氯化钠溶液稀释至100ml,30分钟内滴注完毕,开始要慢。1周2~3次,可根据补铁总量决定。如采用一次性滴注给药方法,右旋糖酐铁500mg(10ml)应稀释至500ml,静脉滴注1~2小时。也可不经稀释肌内注射,每次100mg。
蔗糖铁注射液(Venofer,100mg/支),可以静脉推注和静脉滴注,不建议肌注。静脉滴注时只能用生理盐水稀释,100mg(5ml)稀释于100ml生理盐水中静脉滴注。如为首次使用,可以做过敏试验(也可以不做,由医师决定),即将上述溶液的25ml缓慢滴注,如无反应,即可将剩余剂量在30分钟内输完。也可以不经稀释直接静脉推注,100mg(5ml)至少推注5分钟,每次最大推注剂量为200mg(10ml)。
葡萄糖酸铁钠(62.5mg/支),125mg(10ml)用0.9%生理盐水稀释,静脉滴注1小时。
【预防】
应加强妇幼保健,预防早产,做好喂养指导,提倡母乳喂养,及时添加含铁量及铁吸收率高的辅食品。重视置节育环月经过多的问题。防治寄生虫病,特别是钩虫病。积极治疗慢性出血病灶。对早产儿、妊娠期妇女、胃切除者及反复献血者,应预防性口服铁剂。在高危人群也可推行铁强化食品。
二、慢性病贫血
慢性病贫血(anemia of chronic disease,ACD)是临床上继缺铁性贫血之后的第二大常见的贫血,常继发于慢性感染、炎症和恶性肿瘤等疾病,一般在原发疾病控制不良的1~2个月以后发生,少数见于急性感染或炎症,因为其发病与炎症细胞因子增多密切相关,所以亦称为炎症性贫血(anemia of inflammation)。其特点是血清铁降低,但单核巨噬细胞系统有充足的铁储存。而继发于慢性系统性疾病的贫血是指伴发于慢性系统性疾病的贫血,包括慢性肾脏疾病、肝脏疾病、内分泌疾病等(详见本章第十二节“其他类型的贫血”)。其与ACD的发病机制不同,一般无ACD特征性的铁代谢异常。但在某些疾病的晚期,例如慢性肾脏疾病,存在持续感染和炎症,有免疫机制的参与,也会出现与ACD一样的铁代谢异常。
【发病机制】
炎症性贫血主要的发病机制是炎症性细胞因子增多,如白介素-6、肿瘤坏死因子、干扰素等,导致Hepcidin增多,Hepcidin会阻止铁从肠道吸收(血清铁减低),也会阻止铁从巨噬细胞中释放(储存铁增多,血清铁蛋白增加),从而铁稳态失衡,铁利用障碍,发生贫血。促红细胞生成素(EPO)减少和对EPO敏感性下降也是发病机制之一。自身免疫性疾病发生ACD的患病率为8%~71%,急性和慢性感染发生ACD的患病率为18%~95%。
肿瘤相关性贫血(cancer related anemia)由多因素引起,如肿瘤本身引起的失血、溶血、骨髓侵犯、营养不良等,肿瘤化疗和放疗会导致骨髓抑制,如果这些原因排除后仍有无法解释的贫血,则要考虑肿瘤导致炎性细胞因子释放增多,引起的炎症性贫血。肿瘤发生ACD的患病率为30%~77%。
【诊断与鉴别诊断】
1.国内诊断标准如下
(1)贫血多为轻、中度。
(2)常有感染、炎症、肿瘤等基础疾病。
(3)多数为正细胞、正色素性贫血,也有20%~50%表现为小细胞低色素性贫血,但MCV很少<72fl。
(4)血清铁和总铁结合力均降低,运铁蛋白饱和度正常或降低,血清铁蛋白增高。
(5)红细胞游离原卟啉增多。
(6)骨髓铁染色提示骨髓小粒中巨噬细胞铁颗粒增多,而幼红细胞内铁减少。
(7)EPO水平低于贫血时应有的EPO水平。
2.鉴别诊断
临床上经常会遇到ACD合并缺铁性贫血的患者,当血清铁蛋白<100μg/L时,就要警惕有无ACD合并缺铁的可能;当铁蛋白>100μg/L时可以排除缺铁,诊断为单纯ACD。如果可以同时检测sTfR,就更容易判断有无合并缺铁,sTfR增高,或sTfR/log铁蛋白>2则诊断为合并缺铁。网织红细胞血红蛋白量(CHr)不受炎症等急性时相反应的影响,也可以帮助判断有无合并缺铁,如果CHr<28pg,可以诊断合并缺铁(表16-2-6)。
表16-2-6 慢性病贫血与缺铁性贫血的实验室鉴别指标

注:sTfR:可溶性转铁蛋白受体;CHr:网织红细胞血红蛋白;↓:降低;↑:升高
【治疗】
1.治疗基础疾病
ACD最有效的治疗是治疗基础疾病,包括急性和慢性感染、自身免疫性疾病、肿瘤等。有时基础疾病不能治愈,但治疗有症状的贫血可以提高生活质量和改善预后。
2.输血
一般不需要输血治疗,仅在某些致命的情况下,如合并大量失血的慢性病贫血或Hb<60g/L时需要输血。
3.静脉补铁治疗
在慢性病贫血中补铁需慎重,只有在慢性病性贫血合并明确的缺铁,或EPO治疗造成功能性缺铁时,才需要补铁治疗。铁蛋白≤30μg/L且转铁蛋白饱和度<20%,属于绝对性缺铁,必须补铁。铁蛋白30~800μg/L且转铁蛋白饱和度20%~50%,可能存在功能性缺铁,根据临床需要补铁。铁蛋白>800μg/L或转铁蛋白饱和度≥20%,不存在缺铁,不需补铁。因为小肠吸收铁的功能受抑,所以需要补铁时首选肠道外补铁。用法和剂量参照缺铁性贫血。
4.EPO及其类似物
ACD治疗中EPO的作用已得到公认。常用重组人EPO(rhEPO)制剂有三种:epoetin-α、epoetin-β及darbepoetin(达依泊汀)。开始治疗的指征为Hb<100g/L,治疗的目标值为110~120g/L,不超过120g/L。
EPO常用剂量为100~150U/kg,或10 000U,每周3次;或者30 000~40 000U,每周1次。有反应的患者2周内Hb上升≥10g/L,减量25%~40%维持,如果Hb≥120g/L则暂时停用,Hb下降到120g/L后再以初始剂量的40%开始应用。维持Hb在110~120g/L左右。如果上述剂量应用4~6周后Hb无提高,则EPO剂量加倍,同时根据铁代谢指标,有功能性缺铁时静脉补铁,应用4~6周后仍无反应,则认为EPO治疗无效,停用。
达依泊汀是长效制剂,半衰期是普通EPO的3倍,每2~3周用1次,可以提高患者依从性,初始剂量为每周2.25μg/kg,也可以200μg,每2周1次,或500μg,每3周1次。
EPO治疗后可增加心血管事件、血栓栓塞事件发生,有血栓形成高危因素的患者可以用低分子肝素预防。长期治疗的副作用是高血压,应用抗高血压药物或EPO减量后可以控制血压。应用EPO的一个罕见的并发症是发生纯红细胞再生障碍性贫血,是由于产生了EPO抗体,必须马上停药。
EPO是否促进肿瘤的生长和复发,缩短生存期,存在争议。所以不建议用于乳腺癌、非小细胞肺癌、头颈部肿瘤、淋巴系统肿瘤、宫颈癌等,也不推荐用于接受激素治疗、生物治疗、放疗的肿瘤患者,只适用于正在接受化疗的患者,化疗结束后要及时停用。对于肿瘤相关性贫血是否应用EPO治疗需要事先慎重评估,权衡利弊和风险。
三、铁粒幼细胞性贫血
铁粒幼细胞性贫血(sideroblastic anemia,SA)是一组异质性疾病,由于铁粒幼细胞血红素生物合成途径中某些酶的缺陷导致血红素生成不足,或参与铁代谢途径的线粒体功能缺陷,导致线粒体内铁沉积和铁利用不良,特征性表现为骨髓中存在大量环状铁粒幼细胞,红细胞无效生成,临床表现为低色素性贫血和体内铁负荷过多。
【分类】
根据病因和发病机制,铁粒幼细胞性贫血可分为遗传性和获得性两大类,其中获得性远较遗传性多见。遗传性SA包括遗传性X连锁( ALAS-2 基因突变最常见,其次是 h ABC7 基因突变)、常染色体显性遗传、常染色体隐性遗传和线粒体细胞病等。获得性SA又可分为原发性和继发性两类。原发获得性铁粒幼细胞性贫血是骨髓增生异常综合征的一个亚型,即伴环状铁粒幼细胞的难治性贫血(RARS)。继发获得性铁粒幼细胞性贫血见于铜缺乏(由于吸收不良)或锌摄入过多,维生素B 6 缺乏,铅中毒,酗酒,药物性铁粒幼细胞贫血等。
【诊断】
诊断SA可以根据以下几条标准:
1.贫血为中度到重度,Hb 40~100g/L。红细胞分布宽度增大。白细胞计数正常,血小板计数正常或增高。
2.外周血涂片 遗传性的往往表现为小细胞、低色素性贫血,可以出现较多的嗜碱性点彩红细胞。RARS可以为正细胞,也可以为大细胞性贫血。极少数遗传性SA可以表现为大细胞性贫血,如罕见的骨髓-胰腺综合征(Pearson综合征),其原因为线粒体DNA突变。
3.铁负荷增加 表现为血清铁蛋白增加,血清铁和运铁蛋白铁饱和度增高。
4.骨髓细胞学及骨髓铁染色检查,可发现至少15%以上的幼红细胞出现特征性环状铁粒幼细胞,这是最主要的诊断依据。依据2008年WHO标准,要求幼红细胞内铁颗粒≥5粒,环核1/3周以上才判断为环状铁粒幼红细胞(图16-2-7)。
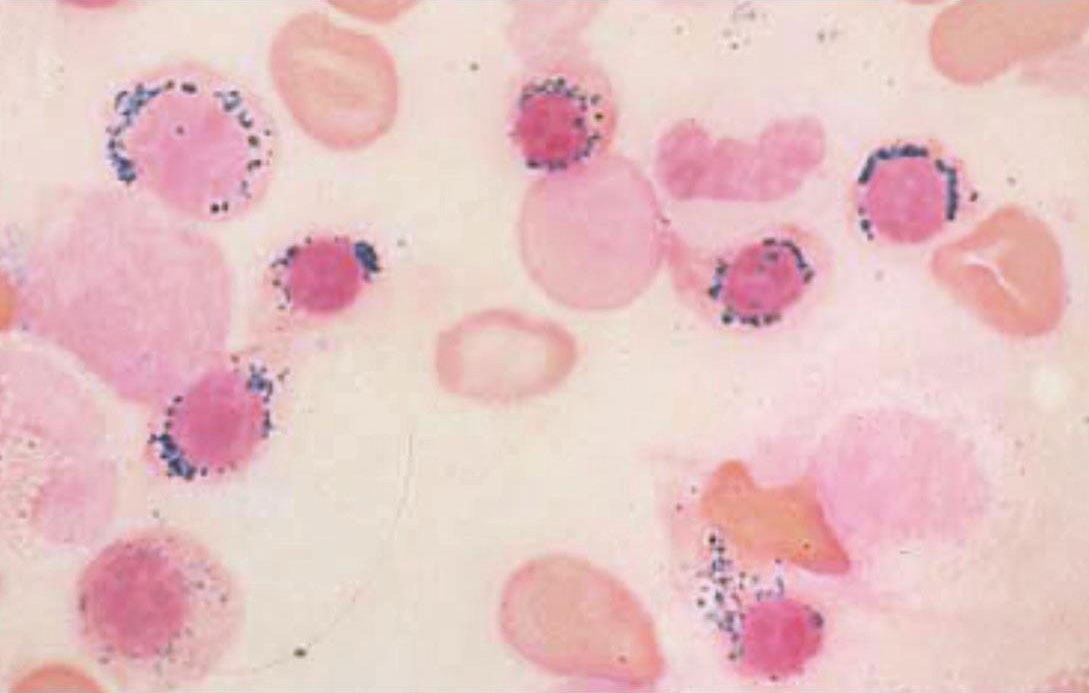
图16-2-7 骨髓铁染色示环状铁粒幼细胞
5.遗传性多有家族史,多数在儿童期发病,少数贫血不严重的也可以到成年才被发现。继发获得性往往有药物、酗酒、中毒等继发因素。
【治疗】
1.治疗基础疾病
如果有明确的酗酒、药物史,停止酒精和药物接触约2周后,骨髓环状铁粒幼细胞可以消失。
2.维生素B 6
对一些营养性SA(如酗酒)和X-连锁的遗传性SA,特别是ALAS-2缺陷的患者,维生素B 6 有一定疗效。所以,凡诊断SA均应该尝试一下该药。剂量为100~200mg/d,口服3个月。如果有效的话,Hb将会上升。长期大剂量治疗有引起周围神经病的危险。
3.输血
如果严重贫血影响到患者的生活质量,则需要输血。对于遗传性SA需要长期输血治疗,不设定目标Hb水平,应定期检查血清铁蛋白,如有铁过载,则需要驱铁治疗。
4.EPO治疗
对于少部分骨髓增生异常综合征RARS亚型,而且血清EPO<500U/L的患者,可以试用2~3个月,无效则停用。
5.异基因骨髓移植
适用于维生素B 6 治疗无效严重型遗传性铁粒幼细胞性贫血和高危型RARS。
主要参考文献
1.马军,王杰军,张力,等.肿瘤相关性贫血临床实践指南(2015—2016版).中国实用内科杂志,2015,35(11):921-930.
2.Camaschella C.Iron-deficiency anemia.N Engl J Med,2015,372:1832-1843.
3.Goldman L,Schafer AI.Goldman's Cecil Medicine.25th ed.Philadelphia:Elsevier Saunders,2016,1068-1071.
4.De Falco L,Sanchez M,Silvestri L,et al.Iron refractory iron deficiency anemia.Haematologica,2013,98(6):845-853.
5.Poggiali E,Migone De Amicis M,Motta I.Anemia of chronic disease:A unique defect of iron recycling for many different chronic diseases.European Journal of Internal Medicine,2014,25(1):12-17.
第五节
巨幼细胞贫血
王小钦
巨幼细胞贫血(megaloblastic anemia)是由于脱氧核糖核酸(DNA)合成障碍所致的一组贫血,主要系体内缺乏维生素B 1 2 或叶酸所致,亦可因遗传性或药物等获得性DNA合成障碍而引起。特点是呈大红细胞性贫血,骨髓内出现巨幼红细胞,粒系、巨核系也可出现巨幼样变。该巨幼细胞易在骨髓内破坏,出现无效性红细胞生成。缺乏维生素B 12 或叶酸所致巨幼细胞贫血是一个逐渐发展的过程,最初是体内叶酸或维生素B 12 储备减少,继之引起叶酸或维生素B 12 缺乏症,最后才引起形态学呈典型表现的巨幼细胞贫血。据人群流行病学调查,叶酸或维生素B 12 缺乏症是全世界最常见的维生素缺乏症。北京大学于2001年在我国河北、无锡、太原地区进行35~64岁健康人群抽样调查,发现叶酸缺乏症患病率南方人为6.2%,北方人38%;维生素B 12 缺乏症患病率南方人为11%,北方人39%。维生素B 12 缺乏症的患病率老年人群尤高,并随年龄而增加。
【维生素B 12 与叶酸代谢】
(一)维生素B 12 代谢
维生素B 12 为含钴的维生素,化学名钴胺(cobalamin,Cbl),仅由某些微生物合成,人体所需的维生素B 12 主要从动物性食物如肉类、肝、鱼、蛋和乳制品等中摄取。成人推荐每天的摄入量为2.4μg,妊娠妇女2.6μg,哺乳期妇女为2.8μg。一般饮食中的供给量已远超过需要量。正常成人体内含维生素B 12 的总量约为2~5mg,其中约1/2贮存在肝内,因此单纯因食物中含量不足而导致缺乏者极为罕见。
食物蛋白中维生素B 12 在胃中经胃酸和胃蛋白酶的作用游离后与结合蛋白(haptocorrin,HC)结合,运送到十二指肠,胰蛋白酶消化HC,释放出维生素B 12 ,维生素B 12 与内因子结合。内因子-维生素B 12 复合物与回肠末端特殊受体(cubam受体)结合,被吸收入回肠黏膜细胞,被运钴胺(TC)转运到血液和组织。血浆中有三种维生素B 12 结合蛋白:运钴胺Ⅰ、Ⅱ、Ⅲ(简称TCⅠ、TCⅡ和TCⅢ)。
血浆中维生素B 12 绝大多数以甲基钴胺的氧化型(Co 3+ )存在,进入细胞内必须还原为Co 2+ 或Co 1+ ,形成具有活性的辅酶形式:甲基钴胺和腺苷钴胺。药用维生素B 12 系氰钴胺,其必须在体内转变为活性形式才能被利用。体内维生素B 12 的主要作用是:①甲基钴胺是甲硫氨酸合成酶的辅酶,催化同型半胱氨酸转变为甲硫氨酸,后者是体内合成蛋白质的必需氨基酸,且S-腺苷甲硫氨酸(SAM)又是体内许多重要酶反应的甲基提供者;②腺苷钴胺是甲基丙二酰辅酶A变位酶的辅酶,促使甲基丙二酰辅酶A转变为琥珀酰辅酶A。
(二)叶酸代谢
叶酸是一种水溶性B族维生素,化学名蝶酰谷氨酸(pteroylglutamic acid)。叶酸在新鲜绿叶蔬菜中含量最多,肝、肾、酵母和蘑菇中也较多。食物烹调、腌制及储存过久等均可被破坏,尤其是加水煮沸损失量尤大。成人推荐每天的摄入量为400μg,妊娠妇女为600μg,哺乳期妇女为500μg。食物中的叶酸以蝶酰多聚谷氨酸的形式存在,要经过小肠中的叶酸多聚谷氨酸水解酶水解成蝶酰单谷氨酸始能吸收。叶酸进入肠黏膜细胞要通过管腔表面的叶酸转运蛋白介导。叶酸在肠黏膜细胞内还原成四氢叶酸(FH 4 ),并且甲基化形成具有活性的甲基-FH 4 ,经门静脉入肝,叶酸的吸收也有肠肝循环。血浆中以5-甲基FH 4 的形式和白蛋白疏松结合运输,通过叶酸受体被摄取进入细胞内,在维生素B 12 依赖的甲硫氨酸合成酶作用下形成四氢叶酸而发挥作用(图16-2-8);亦可再度成为多谷氨酸盐储存,后者可避免叶酸逸出细胞外。叶酸储存于肝细胞内,储存量仅5~10mg,因此营养性巨幼细胞贫血主要由叶酸缺乏所引起。
细胞从血浆中摄取叶酸后,5-甲基FH 4 通过一碳单位的若干传递过程,最后又转变为四氢叶酸。四氢叶酸主要参与一碳单位代谢,后者包括甲基(—CH 3 )、甲烯基(=CH 2 )、甲炔基(—CH=)、甲酰基(O=CH—)和亚氨甲基(HN=CH—)。一碳单位不能游离存在,常与四氢叶酸结合而转运和参加代谢。四氢叶酸缺乏,一碳单位代谢障碍,就会影响DNA合成,引起巨幼细胞贫血。
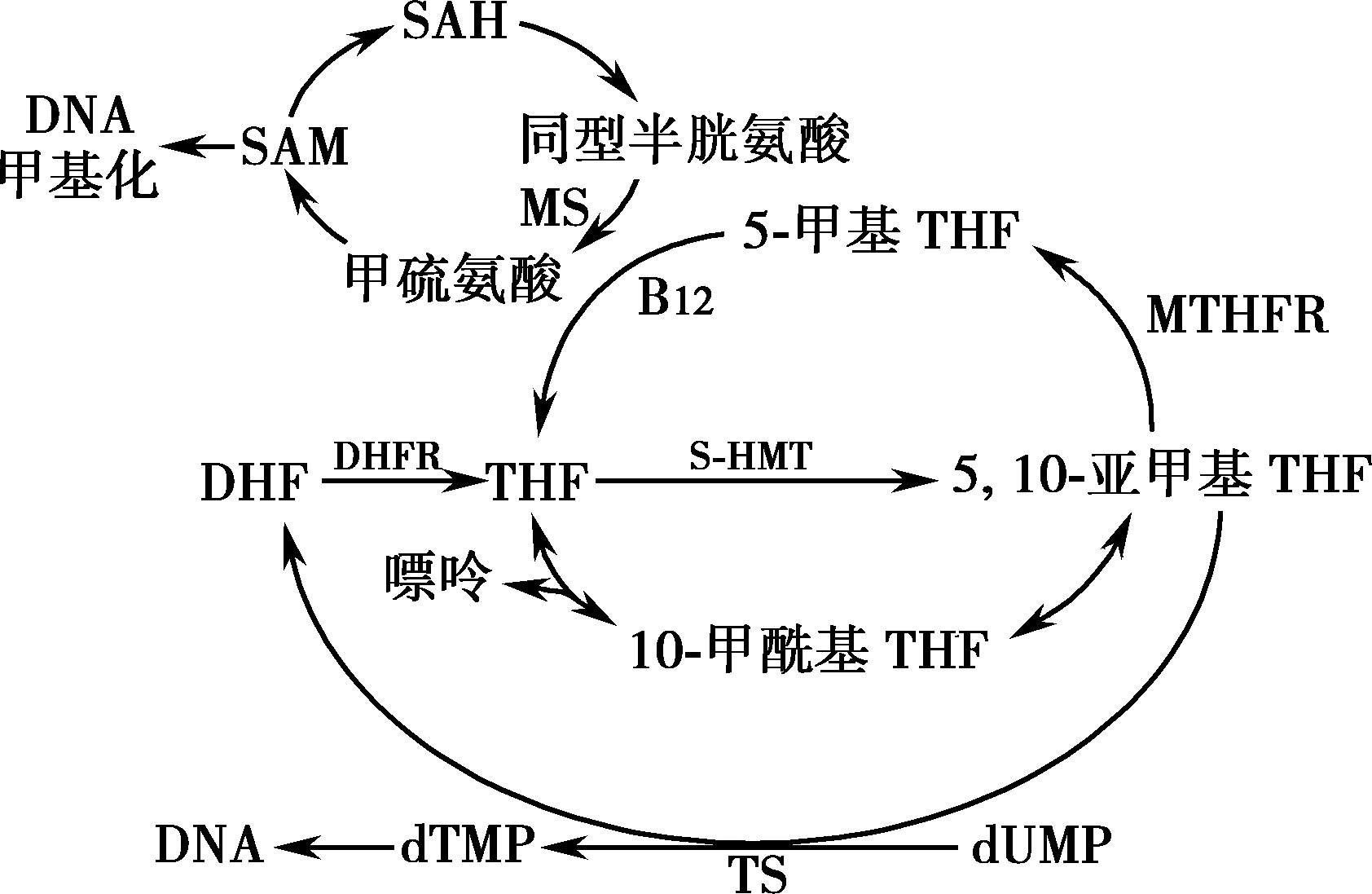
图16-2-8 叶酸代谢
DHF:二氢叶酸;THF:四氢叶酸;SAM:S-腺苷甲硫氨酸;SAH:S-腺苷同型半胱氨酸;MS:甲硫氨酸合成酶;DHFR:二氢叶酸还原酶;S-HMT:丝氨酸羟甲基转移酶;TS:dTMP合成酶;MTHFR:亚甲基四氢叶酸还原酶
【发病机制与病理】
维生素B 12 和叶酸是细胞合成DNA过程中的重要辅酶,维生素B 12 和叶酸缺乏可导致DNA合成障碍。维生素B 12 缺乏导致DNA合成障碍是通过叶酸代谢障碍引起的,维生素B 12 缺乏,细胞内N 5 -甲基四氢叶酸不能转变成其他形式的活性四氢叶酸,并且不能转变为聚合形式的叶酸以保持细胞内足够的叶酸浓度。维生素B 12 和叶酸缺乏,使脱氧尿嘧啶核苷酸(dUMP)转变为脱氧胸腺嘧啶核苷酸(dTMP)发生障碍,使DNA合成速度减慢,过多的dUMP使尿嘧啶掺入DNA,使DNA呈片段状,DNA复制减慢,核分裂时间延长,故细胞核比正常大,核染色质呈疏松点网状,缺乏浓集现象,而胞质内RNA及蛋白质合成并无明显障碍。随着核分裂延迟和合成量增多,形成胞体巨大、核浆发育不同步、核染色质疏松,即所谓“老浆幼核”改变的巨型血细胞。
巨型改变以幼红细胞最显著,幼红细胞形态巨大,核染色质疏松,呈点网状结构。巨原红细胞核仁大而蓝,巨晚幼红细胞核染色质浓集差,核常靠边缘,可呈分叶状,浆内充满血红蛋白。成熟红细胞巨大而厚,常呈卵圆形,缺乏中心苍白区,出现大小不等、嗜多色性或有嗜碱性点彩、卡波环或豪-胶小体等。
巨型改变也见于粒细胞和巨核细胞系,尤以晚幼粒细胞为突出。晚幼粒和杆状核粒细胞形态巨大,核肿大、畸形,核染色质疏松,胞质中颗粒较粗,称巨晚幼粒和巨杆状核粒细胞。分叶核分叶过多,常在5叶以上,甚至达16叶,称巨多叶核粒细胞。巨核细胞体积也增大,核分叶过多,并且核间可不相连接。血小板生成障碍,可见巨大和形态不规则的血小板。
骨髓呈增生象,但血常规显示为全血细胞减少,其主要病理生理改变为无效造血,可有髓内溶血。巨幼细胞和大型红细胞的生存期较正常为短,可出现血清胆红素增高、结合珠蛋白降低、乳酸脱氢酶增高,特别是LDH 1 和LDH 2 (来自幼红细胞)增高。血清溶菌酶增高反映幼粒细胞的破坏。
维生素B 12 还参与神经组织的代谢。维生素B 12 缺乏,S-腺苷甲硫氨酸合成减少,后者导致转甲基反应障碍,造成髓鞘质合成障碍,并且由于腺苷钴胺缺乏,导致大量甲基丙二酰辅酶A及其前身丙酰辅酶A堆积。合成异常的脂肪酸进入髓鞘质,从而导致脱髓鞘病变、轴突变性,最后可导致神经元细胞死亡。神经系统可累及周围神经、脊髓后侧累及大脑。
【病因】
(一)维生素B 12 缺乏症(vitamin B 12 deficiency)
1.摄入不足,需要量增加
即营养性维生素B 12 缺乏症。单纯因摄入不足引起者甚罕见,仅见于长期严格素食者。摄入不足而需要量增加见于妊娠、婴幼儿、溶血性贫血、感染、甲状腺功能亢进及恶性肿瘤等。妊娠期维生素B 12 缺乏可致流产、早产、胎儿宫内发育延迟及神经管发育缺乏。
2.食物蛋白中维生素B 12 释放障碍(食物-钴胺吸收不良综合征)
老年人中维生素B 12 缺乏最常见的原因,约30%~50%的老年人有维生素B 12 储备不足。主要原因与萎缩性胃炎和胃酸缺乏导致食物蛋白中维生素B 12 释放障碍有关,幽门螺杆菌感染及因胃酸缺乏导致小肠细菌过度生长均可加重维生素B 12 缺乏。长期服用剂量较大的H 2 受体拮抗药和质子泵抑制剂也能通过胃酸分泌减少引起维生素B 12 吸收障碍。
3.内因子缺乏
可因胃大部切除或全胃切除,以及自身免疫性破坏(恶性贫血)引起胃壁细胞数量减少、胃酸缺乏,导致内因子缺乏影响维生素B 12 的吸收。全胃切除术后发生巨幼细胞贫血的时间平均为5年(2~10年),约30%~40%的次全胃切除者有内因子缺乏致维生素B 12 吸收不良。罕见病例有先天性分泌无活性的内因子。恶性贫血是西方人群最常见的维生素B 12 缺乏症,主要累及60岁以上人群。恶性贫血是自身免疫性胃炎发展到后期的表现,这种慢性胃炎仅累及胃体,称为A型萎缩性胃炎,引起大量胃壁细胞破坏,从自身免疫性胃炎发展到恶性贫血大约需要20~30年。自身免疫性胃炎患者血清中存在壁细胞抗体,恶性贫血患者存在内因子抗体。内因子抗体有两型:Ⅰ型抗体能阻断维生素B 12 与内因子相结合,故又称为阻断抗体;Ⅱ型抗体能阻止内因子-维生素B 12 复合体与回肠末端Cubam受体相结合,从而阻止维生素B 12 的吸收,故又称结合抗体。
4.小肠疾病引起维生素B 12 吸收障碍
包括胰蛋白酶分泌不足引起HC蛋白降解障碍;Zollinger-Ellison综合征可灭活内源性胰蛋白酶;热带/非热带口炎性腹泻、克罗恩病、小肠淋巴瘤、硬皮病等引起吸收不良综合征都可导致维生素B 12 吸收障碍;末端回肠具有丰富的Cubam受体,如回肠切除过多就会影响维生素B 12 的吸收;小肠寄生阔节裂头绦虫,手术盲袋形成和回肠憩室炎因其中细菌过度繁殖,都可夺取食物中的维生素B 12 ,从而影响人体吸收。
5.药物诱发维生素B 12 缺乏
二甲双胍可抑制内因子和胃酸的分泌,抑制转运维生素B 12 进入肠黏膜细胞;考来烯胺、秋水仙碱和新霉素等均可抑制转运维生素B 12 进入肠上皮。NO 2 可灭活维生素B 12 引起功能性细胞内维生素B 12 缺乏。
6.遗传性维生素B 12 缺乏
见于Cubam受体遗传性缺陷引起Imerslund-Gräsbeck综合征和先天性TCⅡ缺乏症。
(二)叶酸缺乏症(folate deficiency)
1.摄入不足,需要量增加
见于婴儿、儿童及妇女妊娠期和哺乳期,需要量可增加3~10倍。营养不良性主要由于新鲜蔬菜及动物蛋白质摄入不足所致。需要量增加尚见于慢性溶血、骨髓增殖症、恶性肿瘤、甲状腺功能亢进及剥脱性皮炎等。妊娠妇女叶酸缺乏可增加婴儿先天性缺陷发生的危险性。血液透析过程,因叶酸丢失过多,也使叶酸需要量增加。婴儿长期用山羊乳喂养也易引起叶酸缺乏。
2.酗酒和慢性酒精性肝硬化
在美国最常见的叶酸缺乏症来自慢性酒精性肝硬化。
3.肠道吸收不良
如小肠吸收不良综合征、热带口炎性腹泻、短肠综合征、小肠疾病等都可引起叶酸缺乏,并且常常同时有维生素B 12 缺乏和缺铁。
4.药物诱发叶酸缺乏症
叶酸对抗物如甲氨蝶呤、乙胺嘧啶和甲氧苄啶都是二氢叶酸还原酶的抑制剂,导致叶酸利用障碍。柳氮磺吡啶可抑制多聚谷氨酸水解成单谷氨酸,从而影响叶酸的吸收。乙胺嘧啶、柳氮磺吡啶和质子泵抑制剂尚可抑制叶酸转运蛋白,从而抑制叶酸吸收。口服避孕药可增加叶酸分解代谢。抗癫痫药可抑制叶酸吸收。
5.遗传因素引起叶酸代谢障碍
例如叶酸转运蛋白的突变引起叶酸吸收不良,遗传性叶酸代谢酶缺陷等。
(三)维生素B 12 或叶酸治疗无效的DNA合成障碍
包括许多抗代谢药如6-巯嘌呤、氟尿嘧啶、羟基脲及阿糖胞苷等的治疗;某些遗传性疾病如乳清酸尿症、Lesch-Nyhan综合征、亚氨甲基转移酶或N 5 -甲基四氢叶酸转移酶缺乏;尚有维生素B 6 反应性巨幼细胞贫血和维生素B 1 反应性巨幼细胞贫血。维生素B 1 反应性巨幼细胞贫血(Rogers综合征)是一种常染色体隐性遗传性疾病,主要特征是巨幼细胞贫血、糖尿病、感觉神经性耳聋、白细胞和血小板不同程度的减少、骨髓中可见环形铁粒幼细胞。主要是 SLC19A2 基因缺陷,该基因编码维生素B 1 转运蛋白。大剂量维生素B 1 治疗可能有效。
【临床表现与类型】
维生素B 12 和叶酸缺乏的临床表现基本相似,都可引起巨幼细胞贫血、白细胞和血小板减少,以及消化道症状如食欲减退、腹胀、腹泻及舌炎等,以舌炎最为突出,舌质红、舌乳头萎缩、表面光滑,俗称“牛肉舌”,伴疼痛。维生素B 12 缺乏时常伴神经系统表现,如乏力、手足麻木、感觉障碍、行走困难等周围神经炎、亚急性或慢性脊髓后侧索联合变性表现,后者多见于恶性贫血,小儿和老年患者常出现精神症状,如无欲、嗜睡或精神错乱。叶酸缺乏可引起情感改变,补充叶酸即可消失。维生素B 12 缺乏尚可影响中性粒细胞的功能。主要的临床类型有:
(一)营养性巨幼细胞贫血(nutritional megaloblastic anemia)
以叶酸缺乏为主,我国以西北地区较多见,主要见于山西、陕西、河南诸省,常有营养缺乏病史,新鲜蔬菜摄入少又极少荤食,加上饮食和烹调习惯不良,因此常伴有复合性营养不良表现,如缺铁,缺乏维生素B 1 、B 2 、C及蛋白质。本病好发于妊娠期和婴儿期。1/3的妊娠妇女有叶酸缺乏,妊娠期营养不良性巨幼细胞贫血常发生于妊娠中末期和产后,感染、饮酒、妊娠高血压综合征以及合并溶血、缺铁及分娩时出血过多均可诱发本病。婴儿期营养不良性巨幼细胞贫血好发于6个月~2岁的婴幼儿,尤其应用山羊乳及煮沸后的牛奶喂养者,母亲有营养不良、患儿并发感染及维生素C缺乏易发生本病,维生素C有保护叶酸免受破坏的作用。
(二)恶性贫血(pernicious anemia,PA)
系胃壁细胞自身免疫性(毒性T淋巴细胞)破坏,胃黏膜萎缩导致内因子缺乏,使维生素B 12 吸收障碍。好发于北欧斯堪的纳维亚人。多数病例发生在60岁以上,发病率随年龄增高而增高,但也有少数幼年型恶性贫血,后者可能和内因子先天性缺乏或异常及回肠黏膜受体缺陷有关。90%左右的患者血清中有壁细胞抗体,60%的患者血清及胃液中找到内因子抗体,有的可找到甲状腺抗体,恶性贫血可见于甲状腺功能亢进、慢性淋巴细胞性甲状腺炎、类风湿关节炎等,胃镜检查可见胃黏膜显著萎缩,有大量淋巴、浆细胞的炎性浸润。本病和遗传也有一定关系,患者家族中患病率比一般人群高20倍。脊髓后侧索联合变性和周围神经病变发生于70%~95%的病例,也可先于贫血出现。胃酸缺乏显著,注射组胺后仍无游离酸。
(三)药物性巨幼细胞贫血(drug-induced megaloblastic anemia)
这组药物包括前述干扰叶酸或维生素B 12 吸收和利用的药物,以及抗代谢药等。药物性巨幼细胞贫血可分两大组:一组是用叶酸或维生素B 12 治疗有效者,另一组是应用上述药物无效者。
【诊断与鉴别诊断】
(一)确定巨幼细胞贫血
主要依据血细胞形态学特点结合临床表现进行诊断。血常规最突出的表现为大卵圆形红细胞增多,且中央苍白区缩小,中性粒细胞核分叶过多。MCV常大于110fl,MCH常大于32pg。中性粒细胞核分叶过多具有特征性,当血中5叶以上的中性粒细胞超过5%,或找到6叶以上的中性粒细胞,或计算100个中性粒细胞的核叶平均数超过3.5,或5叶以上和4叶以下中性粒细胞的比率超过0.17,均具有诊断价值。重症病例常呈全血细胞减少,网织红细胞减少。因无效造血,血清间接胆红素可轻度升高,血清乳酸脱氢酶升高,其中LDH 1 及LDH 2 明显升高,以前者更为显著。骨髓呈增生象,巨幼红细胞系列占骨髓细胞总数的30%~50%,其中巨原红及巨早幼红细胞可达半数以上,需注意在维生素B 12 或叶酸治疗开始6~24小时后即可找不到典型的巨幼红细胞。中性粒细胞分叶过多要早于巨幼红细胞的出现,粒系巨型变在治疗后的恢复要迟于巨幼红细胞。巨幼红细胞糖原染色阴性。
本病细胞形态学改变具有一定的特征性,但必须注意应和引起全血细胞减少、大细胞性贫血及骨髓有巨幼样改变的疾病相鉴别,特别是骨髓增生异常综合征(MDS)中的难治性贫血、急性髓系白血病中的红血病和红白血病、甲状腺功能减退、肿瘤化疗后及先天性红细胞生成异常性贫血等相鉴别(表16-2-7)。有困难时应作诊断性治疗,即肌内注射维生素B 12 和口服叶酸后观察用药后患者是否有临床症状改善、网织红细胞是否升高、巨幼红细胞形态是否迅速消失以及血红蛋白是否上升,从而达到确诊目的。巨幼细胞贫血时,血清铁、运铁蛋白饱和度、血清和红细胞碱性铁蛋白均增高,如降低则表示有缺铁。
(二)确定维生素B 12 或叶酸缺乏症
主要依据血清维生素B 12 或叶酸测定,以及其代谢产物的测定。
1.血清维生素B 12 测定
常用微生物法及放射免疫法,后者测定方便,为临床常用。血清维生素B 12 <148pmol/L(200pg/ml)可诊断维生素B 12 缺乏。有许多因素可影响血清维生素B 12 测定值,叶酸缺乏、妊娠、口服避孕药、TCⅠ缺乏症、多发性骨髓瘤、大剂量维生素C治疗均可引起假性维生素B 12 缺乏;血清维生素B 12 测定值升高尚见于骨髓增殖症、肝脏肿瘤、活动性肝病、先天性TCⅡ缺乏症及小肠细菌过度繁殖。因此评价血清维生素B 12 测定值的临床意义时应同时测定血清叶酸值。该试验敏感度约65%~95%,特异度约50%~60%,特别对于存在内因子抗体的患者不易正确诊断。
表16-2-7 大红细胞性贫血鉴别诊断
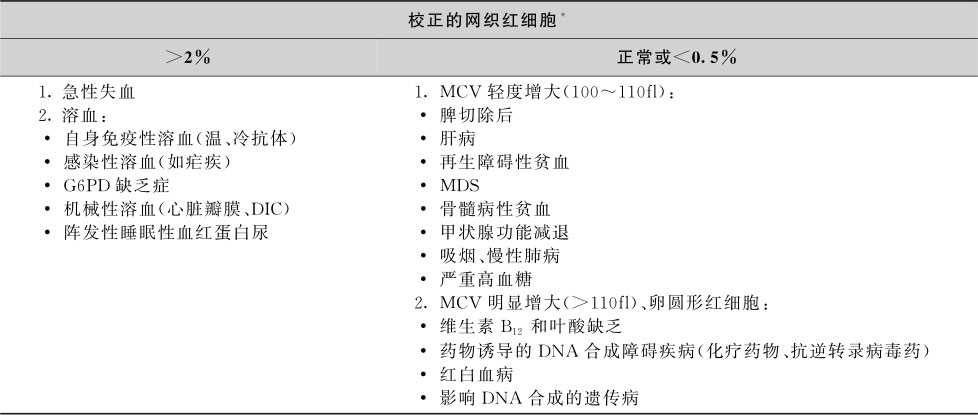
注: * 网织红细胞校正值(%)=网织红细胞(%)×血细胞比容(%)/0.45
2.血清及红细胞叶酸测定
可用微生物和放射免疫法测定,前者较正确,后者方便为临床常用。血清叶酸<6.8nmol/L(3μg/L)或红细胞叶酸<363nmol/L(160μg/L)可诊断为叶酸缺乏。由于血清叶酸水平极易受饮食的影响,而不能反映组织内的叶酸水平,因此要以红细胞内叶酸为准。单独血清叶酸水平减低可见于1/3的住院患者伴厌食者,急性酒精中毒、正常妊娠及使用抗癫痫药的患者。25%~55%酗酒者有血清叶酸浓度降低。
3.血清同型半胱氨酸及甲基丙二酸测定
同型半胱氨酸和甲基丙二酸(methylmalonic acid,MMA)是维生素B 12 和叶酸的代谢产物。同型半胱氨酸转变为甲硫氨酸需要维生素B 12 和叶酸作为辅酶,因此不论维生素B 12 或叶酸缺乏,都可以使血清同型半胱氨酸水平升高。甲基丙二酸CoA转变为琥珀酰CoA仅需要维生素B 12 ,因此维生素B 12 缺乏可使血清MMA水平升高。血清同型半胱氨酸的正常参考值为5~14μmol/L,血清 MMA的正常参考值为70~270nmol/L。如果以血清同型半胱氨酸>21μmol/L,或血清 MMA>400nmol/L为诊断阈值,其敏感度和特异度均可以达到90%以上,但因价格昂贵难以作为常规项目。目前仅用于血清B 12 或叶酸测定难以确定是否有维生素缺乏或是哪一种维生素缺乏时才选用。须注意脱水和肾衰竭可使两者均升高。
(三)确定维生素缺乏的原因
需要借助病史、体检、胃肠道检查及寄生虫检查分析维生素缺乏的病因。疑有恶性贫血则可能需要测定血清内因子抗体和血清壁细胞抗体。
【治疗】
(一)补充治疗
根据缺什么补什么的原则,应补足应有的贮存量。维生素B 12 缺乏可肌内注射维生素B 12 ,最常用为氰钴胺,每次剂量为500~1000μg,开始每天1次,连续1周,后改每周2次,共2周,然后每周1次,共4周;维持量为每月1次,每次1000μg肌内注射,直到病因去除。凡恶性贫血、胃切除者、Imerslund综合征及先天性内因子缺乏者,需终身维持治疗。有神经系统症状的患者治疗剂量要比较大。维生素B 12 的其他制剂也可选用,如羟钴胺(hydroxocobalamin)、甲钴胺(mecobalamin)。由于羟钴胺在组织潴留时间比氰钴胺长,因此可每1~3个月肌内注射1次。
晶体型维生素B 12 亦可口服治疗,但需要较大剂量。因为只有1%~2%的口服维生素B 12 可通过肠道被动弥散吸收。初剂氰钴胺每天1~2mg口服,连续3个月,维持量每天500μg用于摄入不足及食物中钴胺吸收不良的患者,对恶性贫血患者需要每天1~2mg口服维持。
维生素B 12 治疗1~2个月贫血被纠正,6个月左右神经系统症状改善。维生素B 12 缺乏单用叶酸治疗是禁忌的,因会加重神经系统损害。一般不需要输血,给予足量的治疗2~3天后患者状况就会有极大的改善,即使贫血尚未纠正。
叶酸缺乏者可口服叶酸,每日3次,每次5mg,对肠道吸收不良者也可肌内注射亚叶酸钙(甲酰四氢叶酸钙)3~6mg/d,直至贫血和病因被纠正。如不能明确是哪一种缺乏,也可以维生素B 12 和叶酸联合应用。也有认为对营养性巨幼细胞贫血,两者合用比单用叶酸效果为佳。补充治疗开始后1周网织红细胞升高达到高峰,2周内白细胞和血小板恢复正常,约4~6周贫血被纠正。
(二)其他原因导致的巨幼细胞贫血
如果是药物因素导致的,尽可能减量或停药。亚叶酸(5-甲酰基四氢叶酸)可以有效对抗叶酸拮抗药抑制二氢叶酸还原酶的作用,剂量为100~200mg/d。铁粒幼细胞性贫血的巨幼变,可以试用维生素B 6 ,剂量必须达到100mg/d才有效。维生素B 1 反应性巨幼细胞贫血用维生素B 1 治疗,剂量25mg/d,成人反应较差。
(三)病因治疗和其他辅助治疗
应积极去除病因,治疗原发疾患。上述治疗后如贫血改善不满意,要注意有否合并缺铁,重症病例因大量红细胞新生也可出现相对性缺铁,要及时补充铁剂。严重病例补充治疗后血钾可突然降低,因为大量血钾进入新生的细胞内,所以要及时补钾,尤其对老年患者及原有心血管病者。营养性巨幼细胞贫血可同时补充维生素C、B 1 和B 6 。
【预防】
加强营养知识教育,纠正偏食习惯及不正确的烹调习惯。婴儿应提倡母乳喂养,合理喂养,及时添加辅食品。妊娠妇女应多食新鲜蔬菜和动物蛋白质,妊娠后期可补充叶酸。在营养性巨幼细胞贫血高发区,应积极宣传改进食谱。对慢性溶血性贫血、骨髓增殖性疾病或长期服用抗癫痫药者应给予叶酸预防性治疗(1mg/d),全胃切除者应每月预防性肌内注射维生素B 12 1次(100~1000μg)。长期服用质子泵抑制剂或H 2 受体拮抗药的消化道溃疡患者也需预防性口服维生素B 12 (1000μg/d)。素食者应该经常补充维生素B 12 片剂(平均5~10μg/d)。

(资源62) “疑难血液病临床和细胞形态学讨论”之二(病例)
主要参考文献
1.Hao L,Ma J,Stampfer MJ,et al.Vitamin B 12 deficiency is prevalent in 35-to 64-year-old Chinese adults.J Nutr,2007,137(5):1278-1285.
2.Goldman L,Schafer AI.Goldman's Cecil Medicine.25th ed.Philadelphia:Elsevier Saunders,2016,1104-1113.
3.Carmel R.How I treat cobalamin(vitamin B 12 )deficiency?Blood,2008,112:2214-2221.
4.Stabler SP.Vitamin B 12 Deficiency.N Engl J Med,2013,368(21):2041-2042.
第六节
红细胞膜缺陷所致的溶血性贫血
王小钦 林果为
红细胞膜由双层脂质及膜蛋白构成。膜脂质主要由磷脂和未脂化胆固醇以1∶1摩尔比组成。膜蛋白种类繁多,膜蛋白电泳至少有7条以上电泳区带,以分子量从大到小排列命名为带1~带7,其中带1及带2分别为收缩蛋白的α链及β链;带5为肌动蛋白;带2.1为锚蛋白;带3蛋白是红细胞的阴离子交换蛋白;带4.1蛋白功能是加强收缩蛋白与肌动蛋白连接及通过它与血型糖蛋白连接;带4.2蛋白可加强锚蛋白与带3蛋白之间的连接。膜蛋白按其分布和功能分为外周蛋白和内嵌蛋白两大类。外周蛋白中以骨架蛋白为主要成分,构成网络状红细胞膜骨架贴于红细胞内面,包括带1、2、2.1、4.1和5蛋白,其主要功能是维持红细胞形态和变形能力。内嵌蛋白主要为带3蛋白和血型糖蛋白,均嵌入膜中,和红细胞内阴离子转运和能量代谢有关,后者是红细胞的血型标志,并和骨架蛋白相连接,构成膜结构的纵向连接。膜骨架蛋白横向连接是指:膜收缩蛋白(四聚体)-4.1蛋白-肌动蛋白(actin)-膜收缩蛋白(四聚体)。膜骨架蛋白纵向连接有二:①膜收缩蛋白(四聚体的β链)-锚蛋白-带3蛋白;②膜收缩蛋白-4.1蛋白-血型糖蛋白C(图16-2-9)。
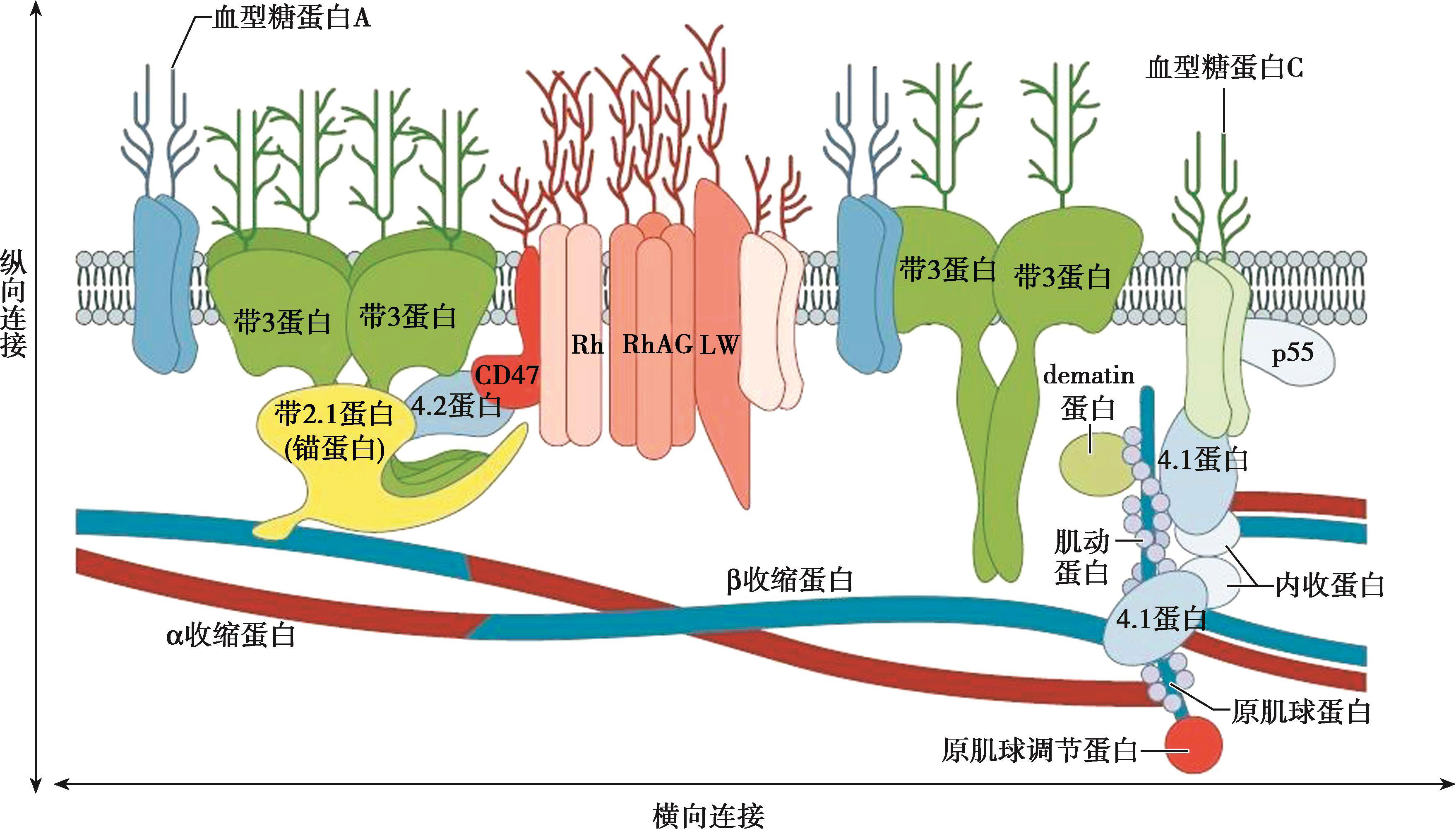
图16-2-9 红细胞膜结构的模式图
注:Rh:Rh血型相关多肽;RhAG:Rh相关糖蛋白;LW:Landsteiner-Weiner 糖蛋白(与血型相关)
遗传性红细胞膜病主要是由于上述这些蛋白的质和量的改变所致。影响红细胞膜不稳定的因素有以下几类:①膜的变形性与胆固醇含量有关,磷脂与胆固醇的比值改变,将出现溶血;②膜骨架结构异常是引起膜不稳定的主要因素;③ATP缺乏时细胞内K + 少,Na + 多,Mg 2+ 也增多;Ca 2+ 与收缩蛋白结合,使红细胞变形能力差,渗透性脆性增加,易于破坏。
一、遗传性球形红细胞增多症
遗传性球形红细胞增多症(hereditary spherocytosis,HS),是一种红细胞膜缺陷引起的遗传性溶血性贫血。其遗传方式有常染色体显性遗传、常染色体隐性遗传及新的突变等,其中75%的病例是常染色体显性遗传。有1/4的HS没有明确的家族史,可能与基因新的突变有关。本病为国内和北欧人遗传性膜缺陷病中最常见者,北欧人群发病率约为1/3000~1/2000。临床主要特征有球形红细胞显著增多,对低渗盐液脆性增加,并有不同程度的黄疸和脾大。脾切除疗效良好。
【病因与发病机制】
HS的基本病变是基因突变,导致膜骨架蛋白缺陷(合成减少或蛋白不稳定)。其分子缺陷主要发生在膜收缩蛋白(α、β链)、锚蛋白、带3蛋白和4.2蛋白,锚蛋白突变占40%~50%,带3蛋白突变占20%,收缩蛋白α与β链突变各占10%,4.2蛋白突变较少。由于收缩蛋白缺乏导致膜脂质缺乏支撑而自动流失,最终使膜表面积丢失,形成球形红细胞。
由于原发性膜缺陷,膜的被动性钠盐流入的通透性增加,水随钠盐而进入细胞内,为了保持细胞内外钠盐浓度的正常比例,就需要产生更多的ATP,以加速钠的排出和钾的摄入。所以球形红细胞的糖酵解率往往较正常红细胞增加20%~30%,以补偿大量ATP的消耗。ATP的相对缺乏使膜上钙-活性ATP酶受到抑制,钙容易沉积在膜上。球形红细胞的直径虽然小于6μm左右,但由于细胞膜变形性和柔韧性减退而被阻留在脾索内,不能通过内皮细胞间空隙(直径仅为3μm左右)进入脾窦。大量红细胞在脾索内滞留过程中,ATP及葡萄糖进一步消耗,代谢缺陷更加剧,终致破坏而溶解。
【临床表现】
HS是一组异质性疾病,可有不同的遗传方式、不同膜蛋白缺陷及不同临床严重度,临床表现多样,诊断困难。典型的表现为贫血、黄疸和脾脏肿大。贫血可轻可重,甚至无贫血。外周血片球形红细胞可多可少,即使无球形红细胞也不能排除HS。黄疸和贫血可不成比例,HS患者可合并Gilbert综合征,该时黄疸深,但无贫血。根据疾病严重度分为以下四种:①无症状携带者临床无溶血征象,但红细胞渗透脆性可增加,有HS基因病变,后代可发生HS;②轻型HS由于骨髓代偿功能好,可无或仅有轻度贫血及脾大,血清胆红素和网织红细胞计数轻度增高,外周血球形红细胞少见;③典型HS自幼年发病,有轻及中度贫血,间隙性黄疸及脾大,有明显的家族史;④重型HS少见,贫血严重,常依赖输血,生长迟缓,面部骨结构改变类似珠蛋白生成障碍性贫血,易出现溶血或再生障碍性危象。人群中以轻型占多数,携带者和轻型HS甚难诊断,只有在临床突发事件,如妊娠、叶酸缺乏、感染等时才出现贫血。凡40岁以下出现胆石症、间歇性黄疸、贫血、新生儿期高胆红素血症等都应怀疑HS。
并发症包括:①溶血危象:最常见,病程呈自限性,一般发生于各种感染所致的单核-巨噬细胞系统功能一过性增强。②再障危象:少见,症状重可危及生命,常需要输血,临床特征为骨髓红系增生低下,网织红细胞计数降低。一般由微小病毒B 19 感染所致。③巨幼细胞贫血危象:当饮食中叶酸供给不足或机体对叶酸需求增加如反复溶血、妊娠等而没有及时补充时可出现。④胆囊结石:超过一半的HS患者有胆红素性胆囊结石症,10~30岁之间发病率最高。⑤其他少见的并发症为下肢复发性溃疡、慢性红斑性皮炎和痛风,脾切除后可痊愈。发育异常或智力迟钝很罕见。
【实验室检查】
(一)血常规
除非有急性发作,贫血一般不重,但危象时血红蛋白可低至30g/L左右。网织红细胞计数增高,一般为5%~20%。当再生障碍危象发生时,红细胞数急剧下降,但网织红细胞反而减少甚至阙如。50%以上的HS患者 MCHC增高,MCV降低,呈小细胞高色素性贫血。红细胞形态单一,体积小,呈球形,细胞中央浓密而缺乏苍白区,细胞直径变短但厚度增加(图16-2-10)。典型小球形红细胞数量可从1%~2%到60%~70%,大多在10%以上(正常人<5%)。20%~25%的HS缺乏典型的球形红细胞。

图16-2-10 外周血涂片示球形红细胞
(二)红细胞渗透性脆性试验(OF)
是测定红细胞在不同浓度的低渗盐水溶液内的抵抗能力,主要受红细胞表面积和体积比值的影响。HS红细胞表面积/体积比值低,渗透脆性增高。正常红细胞开始溶血的生理盐水浓度为0.42%~0.46%,完全溶血为0.28%~0.32%,HS红细胞开始溶血的浓度多为0.52%~0.72%,少数为0.87%。红细胞渗透性脆性试验的灵敏度约66%,约20%~25%患者缺乏典型的球形红细胞,渗透脆性试验可正常,但将患者红细胞孵育24小时后,再进行OF试验,可使灵敏度提高(图16-2-11)。免疫介导的溶血性贫血和其他溶血疾患可能出现假阳性。
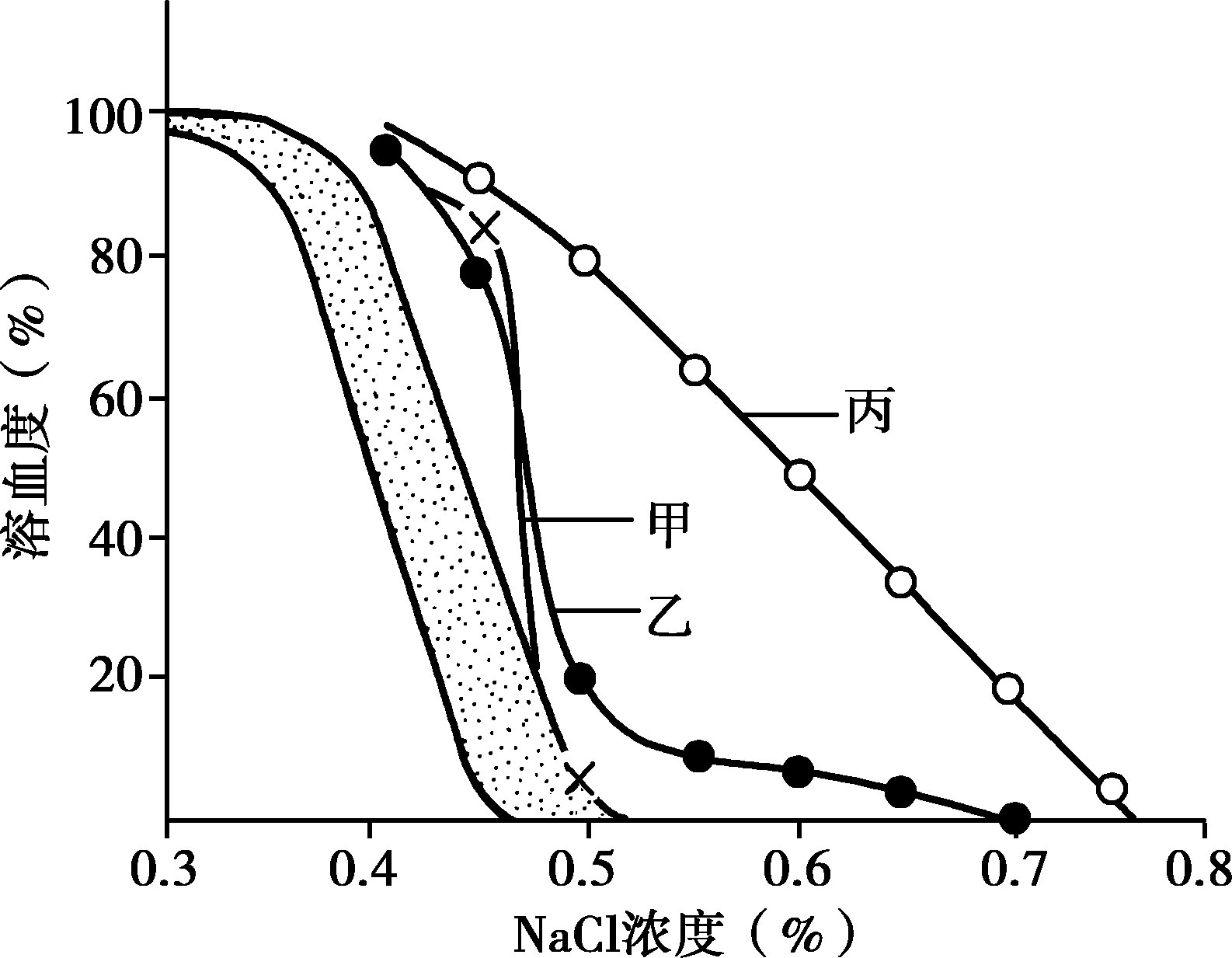
图16-2-11 红细胞渗透性脆性试验的各种不同曲线(遗传性球形红细胞增多症)
甲:轻型,球形红细胞很少,曲线形态接近正常,稍向右偏;乙:球形红细胞稍多,大部分曲线接近正常形态,但尾端则在较高浓度中,呈拖尾曲线;丙:重型,球形红细胞占优势,曲线全偏向右。有点状部分曲线为正常人(图中最左侧)
(三)酸化甘油溶血试验(AGLT)
是测定红细胞在一定浓度的甘油试剂中的溶解速度,用溶解率在50%时的时间来表示(称 AGLT 50 ),正常参考值>290秒,HS的AGLT 50 一般在140秒之内,较正常人显著缩短。作为HS的过筛试验,灵敏度高于OF,但特异度不高,自身免疫性溶血性贫血、慢性肾衰竭、白血病、妊娠妇女可能出现假阳性。
(四)流式细胞仪测定
应用伊红-5-马来酰亚胺(eosin-5-maleimide,EMA)标记红细胞,流式细胞仪测定荧光强度。可反映Rh相关的整合蛋白和带3蛋白的量。HS的荧光强度显著降低。作为HS筛检试验,其灵敏度92.7%,特异度99.1%。
(五)其他
血清间接胆红素增高,多数在(27.36±18.81)μmol/L。红细胞膜蛋白SDS-PAGE分析,80%以上的HS可发现异常,结合免疫印迹法(western blotting),检出率更高。红细胞膜蛋白定量测定,可采用放射免疫法或ELISA直接测定每个红细胞的膜蛋白的含量。应用现代分子生物学技术可在基因水平检出膜蛋白基因缺陷。
【诊断与鉴别诊断】
典型病例具有脾大、黄疸、贫血、球形红细胞增多与红细胞渗透脆性增加,有明确的家族史,诊断即可确立。少数HS需要详细的家系调查或切脾后有效才能确立诊断。极少数HS的诊断需借助红细胞膜蛋白电泳或基因分析。
外周血涂片出现球形红细胞,除了HS外,还见于温抗体型自身免疫性溶血性贫血、新生儿ABO血型不相容性贫血、热损伤、微血管病性溶血、肝脏疾病、梭菌败血症、输血反应的溶血、某些蛇、蜘蛛、毒虫咬伤、严重的低磷酸盐血症等。
一般而言,HS外周血仅有小球形红细胞,其他形态异常的红细胞少见,且球形红细胞形态大小比较均匀一致,而其他溶血性疾病外周血除见到少量球形红细胞之外,常能见到其他形态异常的细胞,且球形红细胞大小不一。HS与自身免疫性溶血性贫血的鉴别:后者在临床更常见,反复的抗人球蛋白试验和对肾上腺皮质激素的治疗反应可资鉴别。
【治疗】
脾切除对本病有显著疗效。但切脾后球形红细胞数不变甚至反而增多。应严格掌握切脾指征,因为切脾后可发生致命的肺炎链球菌败血症,尤其是儿童发生率较高,即使术前接受疫苗接种,术后采用抗生素预防,仍不能完全避免败血症的发生。此外尚有切脾后反应性血小板增多症和肺动脉高压及术后血栓形成的危险。因此,脾切除适用于严重型 HS,中度HS有脾大贫血,如可代偿,也可不切脾。
脾切除的指征:①Hb≤80g/L,网织红细胞≥10%的重型患者。②Hb 80~110g/L,网织红细胞8%~10%的患者具有以下一种情况者才考虑切脾:a.贫血影响生活质量或体能活动;b.贫血影响重要脏器功能;c.发生髓外造血性肿块。③年龄限制:主张10岁以后手术。对于重型HS,手术时机也应尽可能延迟至6岁以上。应提倡腹腔镜切脾,儿童严重型HS也可考虑脾次全切除,以减少术后感染,但易复发。有症状的胆石症才考虑同时切除胆囊。
脾切除失败的原因为:①存在副脾;②因手术中脾破裂而致脾组织植入腹腔形成再生脾;③特殊类型的重型HS;④诊断错误或同时合并其他溶血性疾患如G6PD缺乏症。大多数HS患者应补充叶酸。溶血严重者应给予输血治疗。

(资源63) “疑难血液病临床和细胞形态学讨论”之三(病例)
二、遗传性椭圆形红细胞增多症
遗传性椭圆形红细胞增多症(hereditary elliptocytosis,HE)是一组以外周血红细胞呈椭圆形改变为特征的遗传性溶血性贫血。HE的发生率低于HS,但疟疾高发区其发病率较高。HE的主要发病原因是红细胞膜骨架蛋白的横向连接异常,其次也影响膜骨架的纵向连接,导致膜稳定性降低。
HE可分为4型,即普通型HE、遗传性热变性异形红细胞增多症(hereditary pyropoikilocytosis,HPP)、球形细胞性HE和口形细胞性HE,后者又称东南亚卵圆形细胞增多症(Southeast Asian ovalocytosis,SAO)。其中普通型、球形细胞性HE及SAO为常染色体显性遗传,HPP为常染色体隐形遗传,因此多数病例可追溯到家族史。
普通型HE最常见。杂合子无症状,仅见外周血有椭圆形红细胞。纯合子或双重杂合子按贫血、黄疸、脾大的程度分轻、中、重3型,红细胞形态呈椭圆形和棒形(图16-2-12),OF增高,脾切除可获得不同程度的改善。多数轻型和杂合子携带者无须治疗,但轻型HE在合并感染性脾大时也可诱发显著溶血。

图16-2-12 外周血涂片示椭圆形红细胞
球形细胞性HE具有HS和轻型HE双重特征,红细胞形态呈球状椭圆形和球形,OF增高,临床以中、重度多见,脾大明显,脾切除可获得明显改善。
HPP红细胞形态以小椭圆形、小球形、大量碎裂和不规则异形为特征,MCV明显下降,OF增高,临床中以中、重度多见,脾大明显,切脾可获得部分改善。
SAO在东南亚地区多见,分子病变累及带3蛋白,红细胞形态呈口形样卵圆形,中间有横嵴,红细胞膜僵硬,具有抵抗疟原虫的入侵,OF降低。纯合子可产生致死性溶血,因此仅见杂合子存活者,后者无症状无脾大,无须治疗。
HE的诊断主要依据外周血片椭圆形红细胞的增多,一般占25%~90%。国内学者提出超过25%才有诊断意义。正常人椭圆形红细胞一般不超过5%~15%。HE必须与其他引起椭圆形红细胞增多的疾病,如轻型珠蛋白生成障碍性贫血、缺铁性贫血、巨幼细胞贫血、骨髓纤维化、MDS、红细胞丙酮酸激酶缺乏症加以区别。
三、遗传性口形红细胞增多症
遗传性口形红细胞增多症(hereditary stomatocytosis)是一组罕见的常染色体显性遗传性慢性溶血性贫血。细胞形态学特点为红细胞中心苍白区像一条长孔,类似一个微张的鱼口(图16-2-13)。这类细胞在正常人血片中也可找到,但一般少于4%。口形红细胞增多症的发病机制尚欠清,多数学者仍认为系红细胞膜蛋白质异常,导致红细胞膜阳离子通透性异常。临床上有两型:①水肿型口形细胞增多症,因红细胞膜阳离子通透性异常,导致Na + 和水内流,引起红细胞水肿,MCV升高,OF增加;②脱水型口形细胞增多症,以前称为遗传性干瘪细 胞增多症(hereditary xerocytosis),因红细胞膜阳离子通透性异常,导致细胞内阳离子及水的丢失,引起红细胞脱水,除出现口形细胞外,同时见靶形、棘形细胞,MCHC增加,OF减低。

图16-2-13 外周血涂片示口形红细胞
遗传性口形红细胞增多症应和获得性口型红细胞增多症相鉴别,因为临床上以后者常见。水肿型应和急性酒精中毒或肝病相鉴别;脱水型应和镰状细胞综合征、遗传性球形红细胞增多症和血红蛋白C病相鉴别。
严重水肿型口形细胞增多症脾切除有效,但某些患者在切脾后可发生高凝状态和血栓形成。脱水型脾切除属禁忌,因为具有术后血栓形成的高度危险性。
主要参考文献
1.李津婴,万树栋.溶血性疾病.上海:复旦大学出版社,2008,92-134.
2.Goldman L,Schafer AI.Cecil Medicine.25th ed.Philadelphia:Saunders Elsevier,2016,e1080.
第七节
红细胞酶缺陷所致的溶血性贫血
王小钦
一、概 述
红细胞酶缺陷(erythrocyte enzyme deficiency)所致的溶血性贫血又称红细胞酶病(erythrocyte enzymopathy)。遗传性红细胞酶病是指参与红细胞代谢(主要是糖代谢)的酶由于基因缺陷,导致活性改变而发生溶血的一组疾病。迄今已知共有19种酶缺陷,另一种酶(腺苷脱氨酶)系活性增加所致溶血。红细胞酶按照其在红细胞内的代谢作用可归纳为以下三类:①无氧糖酵解途径中的有关酶;②磷酸戊糖旁路和谷胱甘肽代谢的酶;③参与核苷酸代谢的酶。
成熟红细胞无核,细胞器已全部消失,不能进行核酸和蛋白质合成,也不能通过三羧酸循环以及氧化磷酸化进行糖的有氧氧化供能,且又无糖原储存,因此红细胞所需能量主要来源于血浆中的葡萄糖,作为细胞代谢的主要底物,葡萄糖通过两种途径代谢:糖酵解途径和磷酸戊糖旁路(图16-2-14、图16-2-15),产生红细胞生存所需的ATP和辅酶Ⅰ(NADH)、辅酶Ⅱ(NADPH),保护红细胞免于过早破坏。参与代谢的葡萄糖约90%~95%通过糖酵解途径转化成乳酸,这是成熟红细胞合成ATP的主要途径。糖酵解途径也是红细胞NADH的主要来源。另外大约5%~10%的葡萄糖通过磷酸戊糖旁路代谢,是红细胞NADPH的主要来源。NADPH使红细胞中保持高浓度的还原型谷胱甘肽(GSH),能保护红细胞免受氧化剂的损伤。磷酸戊糖旁路和谷胱甘肽代谢途径有密切联系。
红细胞酶缺陷所致溶血性贫血可分以下三类:
(一)红细胞无氧糖酵解途径酶缺陷所致溶血性贫血
无氧糖酵解途径(embden-meyerhof pathway)是红细胞内ATP、2,3-DPG和NADH的主要来源。每分子葡萄糖经糖酵解最终生成2分子乳酸,净生成2分子ATP,是成熟红细胞获取能量的主要方式;2,3-DPG是红细胞中调节Hb对O 2 亲和力的重要因素;NADH的主要作用是还原高铁血红蛋白,以维持血红素铁的还原状态。
无氧糖酵解需要一系列酶的催化作用。任何酶的缺陷都能影响无氧糖酵解过程的进行,使ATP合成减少,导致红细胞能量缺乏,从而引起红细胞变形能力降低,红细胞形态异常,引起溶血。
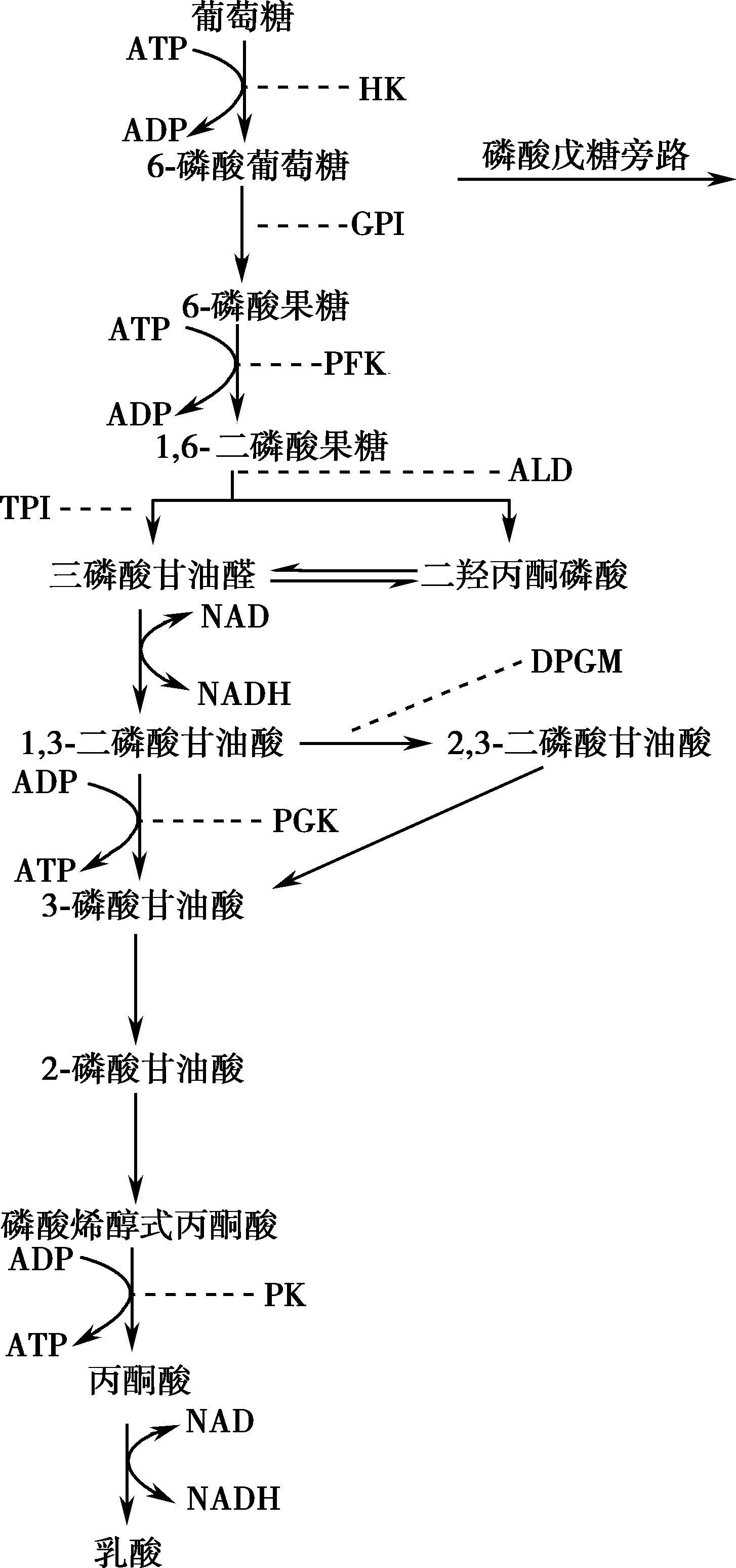
图16-2-14 红细胞无氧糖酵解途径及有关酶缺陷
NAD:氧化型辅酶Ⅰ;NADH:还原型辅酶Ⅰ;DPGM:2,3-二磷酸甘油酸变位酶;HK:己糖激酶;GPI:葡萄糖磷酸异构酶;PFK:磷酸果糖激酶;ALD:醛缩酶;TPI:丙糖磷酸异构酶

图16-2-15 红细胞磷酸戊糖旁路及谷胱甘肽代谢
G6P:6-磷酸葡萄糖;F6P:6-磷酸果糖;NADP + :氧化型辅酶Ⅱ;NADPH:还原型辅酶Ⅱ;GSH:还原型谷胱甘肽;GSSG:氧化型谷胱甘肽
红细胞无氧糖酵解途径酶缺陷疾患包括丙酮酸激酶(PK)缺乏症、葡萄糖磷酸异构酶(GPI)缺乏症、磷酸果糖激酶(PFK)缺乏症、2,3-二磷酸甘油酸变位酶(DPGM)缺乏症,己糖激酶(HK)缺乏症、磷酸甘油酸激酶(PGK)缺乏症等。
按红细胞酶病发病率高低排列,G6PD缺乏症居第一位,PK缺乏症第二位,GPI缺乏症第三位。遗传方式除PGK缺乏症是X连锁隐性遗传(仅男性患病)外,其他均为常染色体隐性遗传。一般纯合子或双重杂合子具有溶血表现,杂合子患者的红细胞含有突变的酶,且活性低于正常,但临床无溶血表现。溶血呈慢性过程,符合先天性非球形红细胞溶血性贫血(CNSHA),常伴脾大。可在婴幼儿或青少年开始出现症状。某些酶缺陷尚可引起其他组织的酶缺陷,如PFK缺乏症可累及红细胞或(和)肌肉,出现肌无力表现。
(二)红细胞磷酸戊糖旁路和谷胱甘肽代谢酶缺乏所致的溶血性贫血
磷酸戊糖旁路(pentose phosphate pathway)又称磷酸己糖旁路(hexose monophosphate shunt),从属于红细胞无氧糖酵解途径,后者所产生的6-磷酸葡萄糖(G-6-P)在葡萄糖-6-磷酸脱氢酶(G6PD)的作用下生成6-磷酸葡萄糖酸(6-PG);接着又在6-磷酸葡萄糖酸脱氢酶催化下变为5-磷酸核酮糖和CO 2 。在这一系列过程中所产生的H + ,使氧化型辅酶Ⅱ(NADP)还原成为还原型辅酶Ⅱ(NADPH)。NADPH是一种重要辅酶,在谷胱甘肽(GSSG)还原酶催化下,使氧化型谷胱甘肽(GSSG)还原为还原型谷胱甘肽(GSH)。GSH是红细胞重要的抗氧化物质,成熟红细胞可合成大量GSH,后者可保护红细胞免受氧化物质的损伤。活性氧包括H 2 O 2 、超氧阴离子(O 2 - )和羟自由基(·OH),这些氧化物质(以 H 2 O 2 为代表)的蓄积就可损伤红细胞的蛋白和脂质。GSH的功能可清除H 2 O 2 对细胞的毒害作用,维持含巯基物质包括Hb、膜蛋白、酶类的还原状态,从而维持红细胞的正常功能和寿命。所以足够量的GSH对保持红细胞的稳定性具有很重要的意义。红细胞磷酸戊糖旁路和谷胱甘肽代谢紧密结合从而保护了红细胞免受氧化物质的损伤。红细胞磷酸戊糖旁路酶缺乏所致溶血性疾患主要是葡萄糖-6-磷酸脱氢酶(G6PD)缺乏症,这是发病率最高的酶病,在遗传性酶病发病率排位中居首位。谷胱甘肽代谢酶异常所致溶血性疾患均罕见,例如γ谷氨酰半胱氨酸合成酶缺陷和谷胱甘肽合成酶缺陷等都可以引起红细胞内GSH减少,导致和G6PD缺乏症类似的临床表现,有轻、中度慢性溶血性贫血,对氧化剂的攻击易感性增高。
(三)红细胞核苷酸代谢酶缺陷所致溶血性贫血
成熟红细胞不能合成嘌呤和嘧啶核苷酸,但可通过补救途径利用磷酸戊糖旁路生成的五碳糖合成核苷酸。临床上有3种红细胞核苷酸代谢酶缺陷所致的溶血性疾患:①嘧啶-5'-核苷酸酶(P5'N)缺陷所致溶血性疾患,发病率居遗传性红细胞酶病的第三位,和遗传性GPI缺陷并列第三;②红细胞腺苷脱氨酶(ADA)缺陷,系唯一因酶活力异常增高导致的遗传性溶血性贫血;③红细胞腺苷激酶(AK)缺陷。后两种少见。
二、葡萄糖-6-磷酸脱氢酶缺乏症
葡萄糖-6-磷酸脱氢酶缺乏症(glucose-6-phosphate de-hydrogenase deficiency)是最常见的红细胞酶病。
【流行病学】
几乎所有的磷酸戊糖旁路缺陷均是因G6PD缺乏所致,这是和溶血性贫血相关的最常见的酶异常,全世界约有4亿人受累。红细胞G6PD遗传缺陷患者遍及世界各大洲,以东半球的热带和亚热带地区为主,不同种族的发生率有很大差异,最高者为土耳其东南部的犹太人(58.2%),也多见于美国及非洲黑种人、意大利和希腊白种人以及西班牙和葡萄牙血统犹太人。因为疟疾流行区G6PD缺陷发生率特别高,所以认为G6PD缺陷可能是逃避恶性疟疾感染的一种优势选择。国内以广西壮族自治区某些地区(15.7%)、海南岛黎族(13.7%)、云南傣族为最多,其次为四川简阳及广东省等。复旦大学附属华山医院采用荧光斑点试验普查上海地区一般人群G6PD缺乏症患病率为0.87%(标化率1.38%)。
【病因】
人类G6PD酶已能提纯,cDNA结构及基因定位都已清楚。G6PD基因定位于X染色体(Xq28)。遗传方式为X伴性不完全显性遗传,具有不同的表现度。男性患者为半合子,由于只有一条X染色体,故酶活力显著缺乏,男性患者与正常女性婚配,所生儿子全部正常,女儿全部为杂合子。女性有两条X染色体,女性杂合子的另一条X染色体等位基因正常,通常溶血代偿良好,而无贫血,但如酶活力显著减低时也可有临床症状。女性杂合子与正常男性婚配,有50%概率遗传给后代,获得突变基因的儿子有临床表现,女儿则50%可能为杂合子。女性纯合子必须父母均有缺陷,可有严重溶血表现,女性纯合子与正常男性婚配,儿子携带该缺陷基因的半合子,女儿均为杂合子。基因突变影响G6PD的编码,迄今已报告160种多种基因突变,大多涉及错义突变,单个氨基酸被置换。国人 G6PD 基因突变类型与国外报道有显著区别,我国最常见的突变型为 G1376T 、 G1388A 和 A95G 。
【病理生理与发病机制】
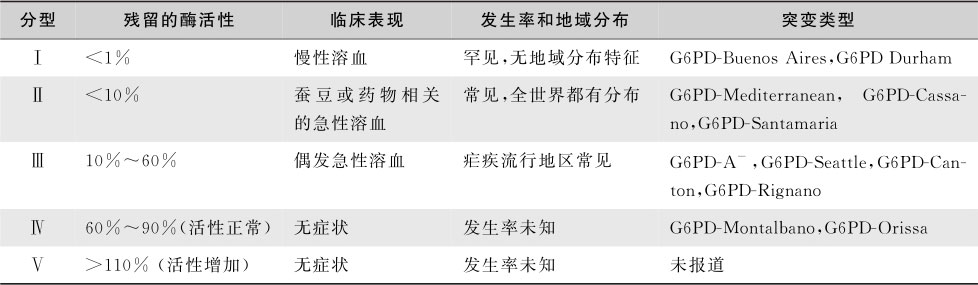
虽然已报道有160种以上 G6PD 基因突变型,导致400种以上的生化变异型,但常见的酶变异型只有少数几种,例如G6PD A-(Gd A- )、G6PD Mediterranean(Gd Med )、G6PD Canton(Gd Canton )、G6PD Seattle、G6PD Union变异型等,其中Gd A- 占绝大多数。WHO根据G6PD酶缺乏程度和溶血严重度将G6PD变异型分为5型(表16-2-8)。Ⅰ型有严重的酶缺陷引起慢性非球形红细胞性溶血性贫血;Ⅱ型也有严重的酶缺陷,通常在蚕豆或药物作用下有间歇性的急性溶血;Ⅲ型酶活力中等,偶发急性溶血,感染和药物是诱因;Ⅳ型酶活力正常,Ⅴ型酶活力反而增高,均无临床意义。
表16-2-8 G6PD缺乏症的WHO分型
G6PD缺陷红细胞,由于不能生成NADPH,GSH显著减少,使红细胞对氧化剂的攻击敏感性增高,Hb的巯基遭受氧化损害,形成高铁血红蛋白和变性Hb,在红细胞内形成Hb沉淀物,并与变性的红细胞膜脂质和膜蛋白形成不可逆的变性珠蛋白小体沉淀在红细胞膜上,称Heinz小体。血涂片中需用活体染色如甲紫染色才能见到。在体内形成的Heinz小体易被脾从循环红细胞中“剔除”,因此脾切除后患者的红细胞中会出现更多的Heinz小体。所谓“咬痕细胞”是指被脾剔除Heinz小体而形成缺失膜表面积的红细胞,可出现于急性溶血发作时的血液循环中。由于红细胞膜脂质和膜蛋白的氧化损伤,影响红细胞膜变形性,更易被脾扣留而破坏。
【临床表现】
G6PD缺乏所致溶血的主要表现为四种临床类型:新生儿高胆红素血症、蚕豆病、先天性非球形红细胞溶血性贫血和药物或感染诱发的急性溶血性贫血。绝大多数G6PD缺乏症无临床表现,在暴露于感染或药物后发生急性溶血,除药物外,感染是诱发溶血的最主要的因素,糖尿病酮症酸中毒也能诱发G6PD缺陷的红细胞破坏。少数G6PD变异型酶活力严重缺乏,在没有感染或药物诱导下呈慢性溶血表现,但慢性先天性非球形红细胞溶血性贫血甚少见。在我国较多见的是蚕豆病和新生儿高胆红素血症及药物、感染诱发的急性溶血性贫血。
(一)新生儿高胆红素血症
包括Ⅰ型和Ⅱ型G6PD缺乏症,常在缺乏明显氧化剂的情况下即发生严重溶血。特别要注意在出生后24小时内发生的黄疸,发病高峰在出生后2~3天。和Gilbert综合征合并存在,黄疸严重而贫血不明显。
(二)蚕豆病(favism)
俗称胡豆黄,是一种由于进食蚕豆后引起的急性血管内溶血性贫血。蚕豆病主要见于意大利、希腊和亚洲。我国四川、桂林、上海、贵州、云南、安徽、广东、北京、江西等地均有报道,国内并不少见。本病因蚕豆中何种成分引起,尚不清楚。蚕豆中含有蚕豆嘧啶、香豌豆嘧啶、异尿咪和伴蚕豆嘧啶核苷,可能是致氧化性溶血的成分。有认为大巢豆素可产生自由基,和发病有关。但同一地区G6PD缺陷者仅部分人发病。患者并不每年食蚕豆均发病,发病程度与食豆量不一定成比例。成人发病显著低于小儿。
患者中绝大多数为1~5岁儿童,3岁以内占病例的70%左右。男性显著多于女性,约占90%以上。本病发生于3~5个月间蚕豆成熟季节。起病多急骤,均在食新鲜蚕豆后几小时(最短2小时)至几日内(一般1~2天,最长15天)突然发作。其严重程度与食豆量无关,有时虽进食1~2粒也会发病。患者贫血多严重,黄疸显著,有重度血红蛋白尿。重症患者尚有酸中毒及氮质血症。实验室检查G6PD活性中至重度缺乏,涉及酶变异型计有30余种。国内所见变异型与药物性溶血相似,Gd Med 对蚕豆敏感。
在高发地区通过普查、普防,国内有些地区发病率已明显下降。患者或家族中有过本病历史者,均应禁食蚕豆,但晒干、煮沸及去皮等处理后似可降低致病机会。多数患者停止食用可自行恢复,严重病例需要输血(应避免输亲属鲜血)及肾上腺皮质激素,并积极纠正酸中毒。
(三)慢性非球形红细胞性溶血性贫血(chronic non-spherocytic hemolytic anemia,CNSHA)
是红细胞酶病溶血性贫血的泛称,其中多数病例是由于G6PD缺陷所致的慢性溶血,具有以下几点特征:①孵育前红细胞渗透脆性试验多不增加;②孵育后自体溶血试验阳性,经加入葡萄糖或ATP可部分纠正;③铁粒幼细胞较多见,尤在脾切除后;④切脾的效果不明显或无效;⑤无异常血红蛋白血症。与药物溶血性贫血不同,自婴幼儿时期起即有轻至中度贫血,可因感染、服药而加重。较肯定相关的感染有伤寒、细菌性肺炎、肝炎等,此外尚有流感、传染性单核细胞增多症、钩端螺旋体病、水痘、腮腺炎等。CNSHA不包括红细胞膜病、血红蛋白病、免疫介导性溶血、PNH等。
脾常肿大,血中无球形红细胞。引起本症的G6PD变异型约计80种以上,酶活性可低至0。国内有G6PD香港型及国外的Gd Med 。一般情况良好或贫血不严重者不需输血。切脾效果大多不佳,所以应严格掌握手术指征。
(四)药物或感染诱发的急性溶血性贫血
药物诱发的G6PD缺陷溶血性贫血以往称为伯氨喹型药物溶血性贫血。除伯氨喹外尚有多种药物(表16-2-9)均具有氧化剂或具有催化血红蛋白氧化变性作用的特性。
表16-2-9 可能诱导G6PD缺陷患者发生溶血性贫血的药物
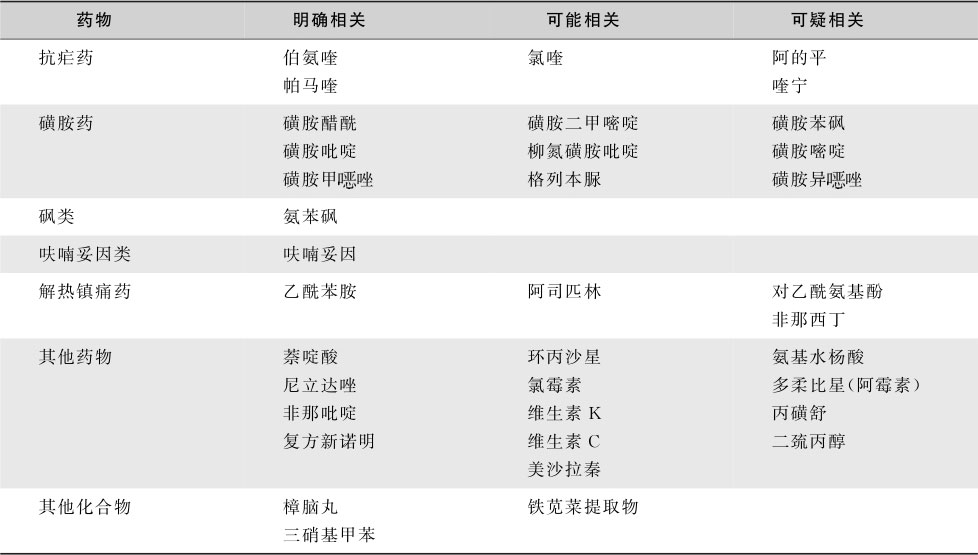
典型表现为在服药后2~3天有血管内溶血发作,一周左右贫血最严重,甚至发生周围循环或肾衰竭。停药后7~10天溶血现象逐渐减退。由于新生红细胞有较高的G6PD活力,因此停用药物后,随幼红细胞的代偿性增多,溶血常为自限性。但也可不自限者,与种族及不同酶变异有关。糖尿病、酸中毒及继发感染,均可加重甚至诱发溶血性贫血。重复用药可再度发作。如果间歇或持续少量用药,可发生慢性溶血。引起此类溶血的变异型有40余种,如Gd A- 、Gd Canton 等。
【实验室检查与诊断】
G6PD缺陷所致的溶血性贫血的诊断除服药史、家族史和临床表现外,主要依靠实验室检查,其方法有以下几种:
1.红细胞G6PD活性测定
直接测定G6PD活性是确定G6PD缺乏最可靠的方法,具有确诊价值,但方法复杂,不适于临床常规应用。本病酶活性为正常的10%~60%,但在急性溶血期及恢复期,G6PD酶活性可正常或接近正常。此时可采用以下方法确定有无G6PD缺乏:全血离心沉淀后取底层红细胞测G6PD活性,如受检者红细胞G6PD活性明显低于正常对照的底层红细胞,可诊断为G6PD缺乏;测低渗处理后的溶血液G6PD活性,如明显降低也可诊断为G6PD缺乏;待急性溶血后2~3个月复测G6PD活性,可以较准确地反映患者G6PD活性。常用WHO推荐的Zinkham法(参考值12.1IU/g Hb±2.09IU/g Hb,37℃)和ICSH推荐的Glock与 Mc Lean法(参考值8.34IU/g Hb±1.59IU/g Hb,37℃),较正常平均值低40%以上有诊断意义。
2.高铁血红蛋白还原试验
正常血红蛋白经亚硝酸氧化为高铁血红蛋白,当红细胞G6PD活性正常时,由于磷酸戊糖旁路形成的NADPH在亚甲蓝的参与下,使高铁血红蛋白还原为亚铁血红蛋白。如果G6PD缺陷,则形成的高铁血红蛋白于一定时间保温条件下还原速度远较正常人慢。高铁血红蛋白为暗红色或棕色,在630nm波长时光密度增高,易与亚铁血红蛋白鉴别。正常人血液孵育4小时为红色,如用分光光度计比色,正常人高铁血红蛋白还原率大于75%。如果孵育后血液为棕色,提示G6PD活性缺陷,如为棕红色可能为G6PD活性缺陷的杂合子,比色法显示酶活性显著缺陷的还原率为30%以下,杂合子为31%~74%。该试验简便,敏感性较高,因此用于过筛试验或群体普查,但有假阳性,可受血红蛋白H(HbH)、不稳定血红蛋白、高脂血症及巨球蛋白血症干扰。
3.氰化物-抗坏血酸盐试验
血红蛋白与抗坏血酸盐接触时能产生过氧化氢,后者将G6PD缺陷的血红蛋白氧化成高铁血红蛋白,产生棕色。本法高度敏感,但也要警惕假阳性。操作方法也不复杂。
4.荧光斑点试验
如果受检标本中G6PD活性正常,则能将试剂中的NADP还原为NADPH,后者在长波紫外线(260~340nm波长)的照射下,发出蓝色荧光。10分钟内出现荧光为正常,10~30分钟间出现为中间缺乏值,30分钟不出现为严重缺乏。如果G6PD活性低于25%即无荧光产生。本试验操作方便,筛检试验中特异性最高。
5.硝基四氮唑蓝纸片法
还原型辅酶Ⅱ(NADPH)通过吩嗪二甲酯硫酸盐的递氢作用,使硝基四氮唑蓝(淡黄色)还原成紫色的甲臜。NADPH生成的量与甲臜产生的量在一定范围内呈线性关系。根据颜色变化,判断有无G6PD缺乏。正常酶活性者,温育后纸片应转为紫色。酶活性缺乏者,纸片仍为红色。酶活性中间值或女性杂合体,纸片颜色介于正常与缺乏中间,为淡紫色。
6.红细胞Heinz小体计数
正常红细胞中不应发现Heinz小体。凡能引起高铁血红蛋白的化学物几乎都能在红细胞内产生Heinz小体,G6PD缺乏以及不稳定血红蛋白病导致溶血时也可发现Heinz小体。化学物中毒后3~4天,Heinz小体可多达30%~50%,G6PD缺乏导致急性溶血后48小时内,Heinz小体也明显增多。先加入乙酰苯肼,37℃孵育后再做甲紫活体染色。红细胞内Heinz小体>5%,有诊断意义。
上述各项试验必须在溶血高峰时操作,必要时2~3个月重复检验。同时检查患者母亲更有意义。此外,尚须注意和获得性G6PD缺乏症鉴别,白血病和MDS可有多种红细胞酶活性改变。复旦大学附属华山医院统计获得性G6PD缺乏症的患病率高达43.1%(标化率42.8%)。
G6PD缺乏症往往有家族史、无法解释的新生儿高胆红素血症、外周血出现咬痕细胞和Heinz小体、在感染或服用药物或食用蚕豆后急性发作,这些均有助于临床诊断,确诊需要进行上述实验室检查。荧光斑点筛选试验和直接测定G6PD活力为最常用的试验。
【治疗】
在没有外源性氧化剂作用的情况下,绝大多数G6PD缺陷者的红细胞表现正常,因此G6PD缺陷本身不需要治疗。防治要点是避免氧化剂的摄入和积极控制感染。轻中度急性溶血者需立即停服相关药物或控制相应的感染,严重溶血者需少量反复输血。由于G6PD缺乏引起的新生儿溶血与一般新生儿溶血的处理基本相同,为了防止神经系统受损,当未结合胆红素>150μmol/L时需要光疗,>300μmol/L时需要输注红细胞进行换血疗法。注意水电解质平衡并保持足够多的尿量,警惕肾衰竭的发生。应用有关药物前,均应询问患者及其家属有无溶血或红细胞G6PD缺陷病史。抗氧化剂(维生素E、硒)疗效不肯定,不推荐切脾治疗。
三、红细胞丙酮酸激酶缺乏症
丙酮酸激酶缺乏症(pyruvate kinase deficiency,简称PK缺乏症)是无氧糖酵解通路中红细胞酶缺陷所致的最常见的溶血性贫血。其发生频率仅次于G6PD缺陷,我国广东地区PK缺乏症基因频率为2.2%,白种人中患病率为1/2万。
【发病机制】
本病为常染色体隐性遗传。纯合子表现明显的溶血性贫血,而杂合子的表现型通常正常。遗传性红细胞PK缺乏症是结构基因突变产生性质异常的酶分子病,现已发现PK基因突变的数目已达100多种,且多数为错义突变,也会发生缺失突变和插入突变。
PK有两种不同的基因( PKM 和 PKLR )编码四种不同的PK同工酶。PK缺乏相关的溶血性贫血由 PKLR 基因突变引起。已知 PKLR 基因突变型超过150种。另一方面结构正常的酶生成减少也可导致PK缺乏。溶血严重度与酶缺乏程度不一定平行。
磷酸烯醇式丙酮酸在PK作用下转化为丙酮酸,同时使ADP转为ATP以供应能量。所以PK缺乏可使ATP产生减少,使红细胞内能量缺失和脱水,发生溶血。
【临床表现】
PK缺乏症临床主要表现为慢性溶血性贫血,溶血严重程度不等,可以轻至完全被代偿,也可以严重到需经常输血。贫血与酶缺陷程度不相平行,因各种变异酶活力各不相同。杂合子酶活性下降50%,可无溶血。溶血主要发生在纯合子。常呈慢性溶血过程,并不受药物或其他氧化剂影响,但可由感染激发。常有脾大。临床过程可因发生再障危象而复杂化,通常由微小病毒感染引起。绝大多数病例溶血过程开始于儿童时期,如有新生儿黄疸,通常需血浆置换,但很少发生核黄疸。10岁以后胆石症发病率上升。脾切除后,溶血常常减轻。
【实验室检查】
1.红细胞PK酶活力测定(正常参考值15IU/gHb±1.99IU/gHb,37℃)为诊断依据 测定时必须除去白细胞,因为PK缺乏症溶血者的白细胞 PK-M 2 基因未受影响,白细胞PK活性是正常红细胞PK活性的300倍。部分红细胞PK缺乏变异型在高底物浓度时,红细胞PK活性接近正常,但在低底物浓度时活性明显降低;也有部分红细胞PK缺乏变异型在实验室检查上主要表现为对其变构因子二磷酸果糖(FDP)的反应异常。因此临床上怀疑红细胞PK缺乏,而红细胞PK荧光斑点筛选试验和常规的PK活性定量检查没有明显异常时,应考虑做低底物(PEP)浓度以及加FDP的PK活性定量测定,同时也要考虑测定红细胞的糖代谢中间产物如2,3-DPG等的含量。年轻红细胞PK活性较高,而衰老的红细胞PK活性降低,因此在诊断红细胞PK缺乏时,应考虑到网织红细胞数量的影响。
2.自体溶血试验 红细胞在患者自己的血清中37℃温育24~48小时发生自发溶血为自体溶血试验阳性,如果先加入葡萄糖或ATP,溶血可得到部分纠正者,为Ⅰ型;溶血不能被葡萄糖纠正而能被ATP部分纠正,则为Ⅱ型。Ⅰ型常由磷酸戊糖旁路中酶的缺陷所致,以G6PD缺乏为最常见;Ⅱ型常由糖酵解途径中酶的缺陷所致,以PK缺乏为代表。自从可以直接测定PK酶活力后,该试验已不再用于PK缺乏症的诊断。
3.红细胞渗透脆性试验正常。
4.红细胞形态 多数病例在脾切除前红细胞形态变化不明显,但脾切除后血涂片中常有典型的少量浓密、刺状的红细胞,但这并不是该病所独有。
【诊断】
直接测定红细胞PK酶活性、测定PK酶的底物和产物的浓度以及在基因组或cDNA水平测定 PK 基因的特异性突变,是诊断红细胞PK缺乏的主要依据。红细胞内ATP含量测定和红细胞形态变化不能作为诊断的依据。
本病需与遗传性球形红细胞增多症、G6PD缺乏症及血红蛋白病相鉴别。骨髓增生异常综合征和白血病可有获得性红细胞PK缺乏,须注意鉴别。
【治疗】
除严重贫血外一般不需输血。输血依赖者可以脾切除,有一定疗效,为了减少脾切除后的并发症和死亡率,一般>5岁后才实施。慢性溶血者需补充叶酸、维生素B 12 。反复输血者要同时去铁治疗。多数病例可活到成年,仅少数死于严重贫血。
四、红细胞嘧啶-5'-核苷酸酶缺乏症
红细胞嘧啶-5'-核苷酸酶(P5'N)缺乏症(pyrimidine-5'-nucleotidase deficiency)是引起遗传性非球形红细胞溶血性贫血的常见病因之一,其发生率仅次于丙酮酸激酶缺陷,最早由Valentine等于1972年报道,国内外报道已有60余例,国内曾报告2例,其遗传方式为常染色体隐性遗传,编码基因位于7p15-p14,患者自幼得病,贫血明显,伴巨脾,外周血嗜碱性点彩红细胞显著增多,多合并智能障碍。具有杂合性生化缺陷的患者家属常无血液学症状,因而不易发现。切脾后症状可部分改善。
主要参考文献
1.Cappellini MD,Fiorell G.Glucose-6-phosphate dehydrogenase deficiency.Lancet,2008,371(9606):64-74.
2.Ronquist G,Theodorsson E.Inherited,non-spherocytic haemolysis due to deficiency of glucose-6-phosphate dehydrogenase.Scand J Clin Lab Invest,2007,67(1):105-111.
3.Tavazzi D,Taher A,Cappellini MD.Red blood cell enzyme disorders:an overview.Pediatric Annals,2008,37(5):303-310.
4.Manganelli G,Fico A,Martini G,et al.Discussion on pharmacogenetic interaction in G6PD deficiency and methods to identify potential hemolytic drugs.Cardiovasc Hematol Disord Drug Targets,2010,10(2):143-150.
第八节
血红蛋白病
翟晓文
一、概 述
血红蛋白病(hemoglobinopathy)是由于血红蛋白分子结构异常(异常血红蛋白病),或由于一种或一种以上珠蛋白肽链不能合成或合成不足,但缺失或不足的珠蛋白肽链一级分子结构正常(珠蛋白生成障碍性贫血,原称地中海贫血及海洋性贫血)所引起的一组遗传性血液病。临床可表现为溶血性贫血、高铁血红蛋白血症或因血红蛋白氧亲和力增高或减低而引起组织缺氧或代偿性红细胞增多所致发绀。
【人类血红蛋白的组成和结构】
人类血红蛋白是一种结合蛋白,由珠蛋白和亚铁血红素组成,分子量64 400,血红素由原卟啉与亚铁原子组成。每一个血红蛋白有两对珠蛋白肽链,一对是α链,包括α与ζ2种肽链,由141个氨基酸残基构成,含较多组氨酸,在运氧中具有重要生理作用。另一对是非α链,有β、γ、δ及ε4种肽链。ζ、ε、α与γ链,分别组成胚胎早期(妊娠3个月以内)血红蛋白、Hb Gower1(ζ 2 ε 2 )、Hb Gower2(α 2 ε 2 )、Hb Portland(ζ 2 γ 2 )。β链含146个氨基酸残基,β93半胱氨酸易被氧化产生混合二硫化物及其他硫醚类物质,可降低血红蛋白稳定性。δ链亦由146个氨基酸残基组成,仅10个氨基酸与β链不同。由于δ链中第22位丙氨酸置换了β22谷氨酸,第116位精氨酸换了β116组氨酸,因此δ链的正电荷大于β链,Hb A2(α 2 δ 2 )等电点升高,电泳时靠近负极。γ链虽由146个氨基酸组成,但与β链有39个氨基酸不同,且含有4个异亮氨酸,为α、β与δ链所缺如,因此可用分析异亮氨酸方法测定Hb F(α 2 γ 2 )含量。初生时 Gγ与Aγ的比例是3∶1,儿童和成人两者之比为2∶3。每一条肽链和一个血红素连接,构成一个血红蛋白单体,人类血红蛋白是由两对(4条)血红蛋白单体聚合而成的四聚体。人类血红蛋白中珠蛋白结构略有不同,但血红素相同。
血红蛋白的四级结构:由氨基酸顺序排列的肽链结构称为血红蛋白的一级结构。肽链中的氨基酸可分为亲水的极化氨基酸(其侧链为羧基、氨基),与非极化的氨基酸(其侧链是芳香族)。肽链中的各种氨基酸的侧链相互拉紧形成α螺旋,螺旋形节段间由短而非螺旋形节段相连。螺旋形节段从N端至C端分别以A-H表示(图16-2-16),α类肽链包含7个螺旋(无D螺旋),非α类肽链包含8个螺旋片段。非螺旋形节段用AB、CD等表示,称为血红蛋白二级结构。血红素的铁原子有6个配位键,第5个配位键结合在肽链F段第8位氨基酸上,第6个配位键结合氧,并间接结合在肽链E段的第7位氨基酸上。肽链围绕血红素为中心,构成内外两层螺旋状蛇形盘曲的三维空间结构,称为三级结构(图16-2-16)。亲水氨基酸分布于外层,使血红蛋白能溶于水而不致沉淀;疏水氨基酸分布于内层,使水分子不能进入血红素腔内部,避免血红素的Fe 2+ 氧化为Fe 3+ 。四个血红蛋白单体(肽链三级结构加血红素),按一定的空间关系结合成四聚体,如Hb A(或Hb A 1 ,α 2 β 2 )、Hb A 2 (α 2 δ 2 )及 Hb F(α 2 γ 2 ),称异质型四聚体;由两对同样的三级结构血红蛋白单体结合成的四聚体,如Hb H(β 4 )及Hb Bart(γ 4 ),称为同质型四聚体。以上所述四聚体为血红蛋白四级结构。综上所述,血红蛋白与分子的外表结构必需完整,带有负电荷,α、β链结合部位要固定,包围血红素腔的氨基酸顺序排列需完整,否则血红蛋白就不能维持分子结构稳定性及正常运氧生理功能,并易遭破坏。
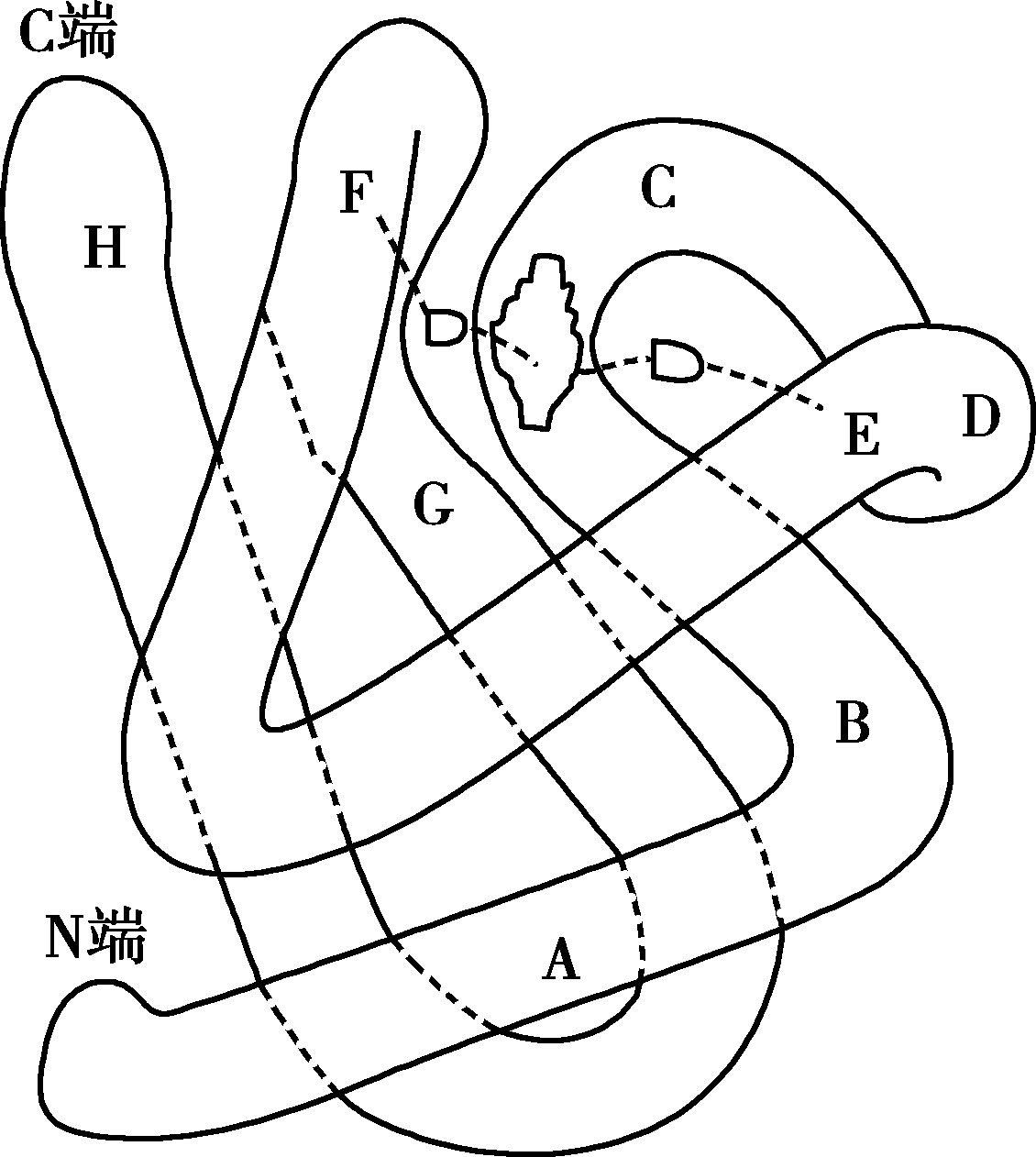
图16-2-16 血红蛋白三级结构示意
肽链N~C端折叠成8个螺旋节段(A~H),螺旋节段由非螺旋节段(AB~GH)相连,血红素位于中心与F 8 E 7 组氨酸相连,构成内外两层螺旋状蛇形盘曲的三维空间结构
【血红蛋白种类和异常血红蛋白】
正常人出生后有三种血红蛋白:①血红蛋白A(Hb A),由一对α链和一对β链组成(α 2 β 2 ),占正常成人及6岁以上儿童血红蛋白总量的90%以上。胚胎2个月时Hb A即有少量出现,初生时占10%~40%,出生6个月后即达成人水平。②血红蛋白A 2 (Hb A 2 ),由一对α链和一对δ链组成(α 2 δ 2 )。自出生6~12个月起,占血红蛋白的2%~3%。③胎儿血红蛋白(HbF),由一对α链和γ链组成(α 2 γ 2 ),初生时占体内血红蛋白的70%~90%,以后渐减直至生后6个月其含量降至血红蛋白总量的1%左右。血红蛋白的不同肽链是由不同的遗传基因控制的,α链基因位于第16号染色体短臂,β、δ、γ链位于第11号染色体短臂,呈连锁关系。
珠蛋白基因突变而致肽链的单个或多个氨基酸替代或缺如,导致珠蛋白分子结构改变,称为异常血红蛋白。若α珠蛋白基因的缺失或缺陷,可导致α珠蛋白链合成减少或缺乏,称为α珠蛋白生成障碍性贫血。若β珠蛋白基因缺陷,可导致β珠蛋白链合成减少或缺乏,称为β珠蛋白生成障碍性贫血。全世界范围内经结构分析证实的异常血红蛋白日益增多,但不到1/3的异常血红蛋白会产生临床症状。世界卫生组织估计,全球约有1.5亿人携带血红蛋白病基因,并已将血红蛋白病列为严重危害人类健康的6种常见疾病之一。主要集中在热带和亚热带地区,好发于地中海沿岸、美国黑人人群、北非、东南亚和印度次大陆等地区,世界上有4.83%的人口携带珠蛋白变异基因。在我国以广西、广东、海南等省发病率较高,其中广西人群中地中海贫血基因携带率为12.22%~23.02%。
【分子遗传学】
血红蛋白病的分子遗传学变化大致可归纳为以下数类:
1.单个碱基替代
由于遗传密码中单个碱基替代,导致由该碱基决定的氨基酸发生相应的变化,形成肽链中单个氨基酸置换的异常血红蛋白,例如Hb S、Hb C等。目前发现的异常血红蛋白中,以本类型最多见,约占90%。
2.终止密码的突变
因终止密码的变异使珠蛋白肽链不在正常的位置终止,导致肽链延长或缩短,如Hb Mc-KeesRock的β链第145位氨基酸的碱基由UAU变为UAA(终止密码),使β链提前结束,仅含144个氨基酸。
3.移码突变
如正常血红蛋白肽链遗传密码中,嵌入或缺失1~2个碱基,使正常三联密码子碱基成分发生改变,如Hb Tak为β链第147位终止密码UAA前插入AC,使UAA→Thr(苏氨酸),而致β链延长至第157位氨基酸,比正常β链多11个氨基酸。
4.密码子缺失或插入
生殖细胞减数分裂时,染色体发生错配或不等交换,形成两种珠蛋白基因。一种失去一部分密码子,合成缺失部分氨基酸的肽链,如Hb Lyon的β链第17~18位缺失赖氨酸、缬氨酸,另一条染色体单体上却嵌入了相应密码子,合成插入部分氨基酸的肽链。β珠蛋白基因和δ珠蛋白基因大片段缺失,可致遗传性胎儿血红蛋白持续存在综合征,或δβ珠蛋白生成障碍性贫血。
5.融合基因
减数分裂时,不同珠蛋白基因之间发生不等交换,合成融合链的异常血红蛋白,如δ链和β链基因错误联合,产生不等交换,形成融合δβ(Hb Lepore)和βδ(Hb反Lepore)。
6.启动子突变
启动子区有单个核苷酸被置换,使启动子功能降低。如β珠蛋白基因启动子在内的小片段缺失,可导致β珠蛋白生成障碍性贫血,表现为Hb A 2 显著升高,可能因主要的转录因子和基因座控制区(LCR)在β珠蛋白基因启动子缺失情况下与δ珠蛋白基因启动子相互协同作用所致。
7.剪接突变
β珠蛋白基因第24~27位密码子突变易发生剪接位点激活,如β24位点GGT→GGA突变(均编码甘氨酸)不引起氨基酸替代,但可改变剪接过程,导致β珠蛋白生成障碍性贫血。天然剪接位点或内含子中的重要位点发生点突变会减少或抑制剪接加工,导致珠蛋白生成障碍性贫血。
【血红蛋白病的种类和分子机制】
血红蛋白病种类繁多,临床症状多样化,但归纳其结构变异所导致功能异常,大致分为以下数类:
1.因分子内部氨基酸替代所产生的异常血红蛋白血红蛋白分子内部为非极性氨基酸,如被不同理化性质的氨基酸替代,会影响分子的构型和稳定性,此类异常血红蛋白包括血红蛋白M(Hb M)、不稳定血红蛋白(UHb)和氧亲和力改变的血红蛋白。
(1)Hb M:肽链中与血红素铁原子连接的组氨酸被酪氨酸所替代,最常见的是E7或F8的组氨酸为酪氨酸所替代,酪氨酸酚基上的氧与血红素的铁原子构成离子键,使铁原子呈稳定的高铁状态,影响血红蛋白的正常释氧功能,使组织供氧不足,出现发绀及红细胞增多。高铁血红素并易与珠蛋白链分离,使血红蛋白分子结构不稳定而发生溶血。
(2)UHb:肽链中与血红素紧密结合的氨基酸发生替代或缺失,影响肽链的立体结构或减弱与血红素的结合力,形成UHb分子。水易进入血红蛋白腔内,使亚铁血红素氧化为高铁血红素;β链第93位半胱氨酸的硫氢基被氧化,产生硫化物,形成硫化血红蛋白,使珠蛋白链与血红素分离。游离珠蛋白链在37℃即不稳定,四聚体易解离为单体,在红细胞内聚集沉淀,形成包涵体,使细胞膜僵硬,通过微循环时往往导致膜部分丧失,最终变为球形红细胞,在脾阻留而破坏。
2.因分子外部氨基酸替代所产生的异常血红蛋白种类很多,一般均对分子构型、功能和稳定性没有明显影响。
(1)Hb E是β链第26位谷氨酸被赖氨酸替代,因谷、赖两种氨基酸理化性质相同,其替代位置虽在α 1 β 1 接触面上,但对血红蛋白分子的稳定性和功能影响不大。这类异常血红蛋白中少数可产生溶解度改变。
(2)Hb S和Hb C均由于其分子外部形状或电荷改变,缺氧时溶解度降低。Hb S聚合为螺旋状体,扭曲成镰刀形纤维;而Hb C聚合成一种副结晶,两者均使细胞膜变硬,难以通过微循环,丧失部分红细胞膜,形成球形红细胞,在脾窦内阻留溶破。
(3)β珠蛋白生成障碍性贫血患者,过剩的α肽链形成多聚体,引起红细胞膜损害,致使大量幼红细胞无效生成。
(4)α珠蛋白生成障碍性贫血,过剩的β及γ链形成Hb H(β4)或 Hb Bart's(γ4)。Hb H是一种不稳定血红蛋白,Hb H包涵体结合在红细胞膜上,使膜对阳离子通透性发生改变,钾盐与水逐渐从红细胞内渗至胞外。缺钾红细胞寿命缩短,易在单核-巨噬细胞系统被破坏,导致溶血。Hb Barts对氧亲和力增高,造成组织缺氧。
【诊断】
本病分布因地域、种族而异,应详细询问患者国籍、籍贯、民族,临床有无黄疸、贫血、肝脾大、生长发育迟缓或发绀、红细胞增多,家系中有无同样病史患者。实验室检查包括网织红细胞计数、血细胞比容、周围红细胞形态及红细胞脆性试验,了解有无低色素、小细胞性贫血。上述检查提示有血红蛋白病可能,应对患者及其家系做相关实验室检查进一步确诊。
目前已建立先进的基因诊断技术,对血红蛋白病进行基因诊断和产前诊断:
1.常用基因诊断方法 包括抽提全血、干纸片血、羊水细胞、绒毛细胞DNA做DNA点杂交,适用于诊断基因缺失的遗传病,如α珠蛋白生成障碍性贫血患者α珠蛋白基因不同程度的缺失。
2.限制性内切酶酶谱法 适用于诊断基因突变改变了限制酶切点或DNA缺失而改变酶解片段大小长短的遗传病。
3.限制性内切酶片段多态性分析(RFLP)RFLP按孟德尔方式遗传,如某种遗传病基因与特异的RFLP紧密相连,即可将这一多态片段作为“遗传标记”,通过RFLP连锁分析推测该家庭成员和胎儿是否携带遗传病基因,RFLP连锁分析适用于诊断已有先证者的单基因遗传病。
4.寡核苷酸杂交 是一种直接基因诊断技术,对于基因突变部位的碱基序列已查明的遗传病,均可以直接检测和鉴定其突变的基因。
5.聚合酶链反应(PCR)DNA体外扩增 此种高效DNA分析技术可以直接通过PCR产物的电泳分析技术进行基因诊断,适用于诊断基因缺失或部分DNA缺失所致的遗传病。RNA诊断检测基因能否转录,转录物(mRNA)是否正常及转录效率的高低等。应用反转录-聚合酶链反应(RT-PCR),不仅解决了RNA的稳定性问题(通过反转录酶将mRNA转换成DNA),且十分灵敏。经氨基酸或基因克隆测序,了解珠蛋白基因病变。
6.对非缺失型突变基因可结合限制酶切位点的改变,如与RFLP位点相连锁,则可用限制酶消化PCR扩增产物,直接电泳分析,不需应用基因探针进行分子杂交,大大简化实验操作,使基因诊断可在半天内完成。
【预防与治疗】
在此病高发地区及患者家系中,务必行婚前检查、遗传咨询及血红蛋白病筛查工作,宣传近亲结婚的危害性,劝阻双方均为本病基因携带者婚配;对高危家系应作产前诊断,早期发现重型胎儿,劝其终止妊娠。对于高发地区已应用RFLP连锁分析,间接检测β珠蛋白生成障碍性贫血基因,或应用人工合成的寡核苷酸探针进行杂交,直接检测突变基因等基因诊断技术
二、珠蛋白生成障碍性贫血
又称海洋性贫血或地中海贫血(thalassemia),因一条或多条珠蛋白链合成速率缺陷导致珠蛋白链合成的不平衡,致无效红细胞生成,它是一组遗传异质性溶血性贫血。珠蛋白生成障碍性贫血是人类最常见的单基因疾病,按受抑制的肽链不同而区分为α、β、δ、δβ和γβ珠蛋白生成障碍性贫血等。α及β珠蛋白生成障碍性贫血是临床上最常见的两种。遗传性胎儿血红蛋白持续存在(hereditary persistence fatal hemoglobin,HPFH)综合征是指患者生后不能将γ链合成转变为β链的合成,而致红细胞内高浓度HbF持续存在。
(一)α珠蛋白生成障碍性贫血
亦称α地中海贫血(α-thalassemia),是一组常染色体隐性遗传病,它是由α珠蛋白链合成减少或缺失而致病,是我国长江以南各省发病率最高,影响最大的遗传病之一,球蛋白链的不平衡导致溶血和红细胞生成障碍,其携带者无症状,不需要治疗。中间型α地中海贫血或血红蛋白H病导致溶血性贫血。重型α地中海贫血的Bart血红蛋白常导致致死性胎儿水肿。
1.流行病学
在世界人口中约5%有珠蛋白变异,其中有1.7%的有α地中海贫血症状。男女地中海贫血患病率一样,新生儿中的发病率约4.4/10 000。α地中海贫血常发生在非洲和东南亚人群中,β地中海贫血常发于地中海、非洲和东南亚人群。我国长江以南的广大地域为该病的高发区,尤以广西、广东和海南三省(区)最为严重,这些地区人群中的重型地中海贫血(包括Hb H病)的发生率为1.2‰~8.1‰,其中α 0 地中海贫血发生率为8.5%,新生儿携带率为0.23%。因此α地中海贫血是我国南方地区为常见的溶血性疾病,在北方地区属少见疾病。
2.分子机制和基因型
α地中海贫血是α珠蛋白链合成减少或缺失,导致β珠蛋白链过剩。α珠蛋白链产物由1条16号染色体上的2个基因控制。产物的缺乏常由1个或多个基因缺失所致。单基因缺失导致α地中海贫血携带者,血液学检测正常,临床无表型。2个基因缺失导致小红细胞症和无贫血的轻度α地中海贫血。3个基因缺失导致Hb H病(中度α地中海贫血),有小红细胞性贫血、溶血和脾大等症状。4个基因缺失导致Hb Bart's,重度α地中海贫血的Hb Bart's常有致死性胎儿水肿。
α地中海贫血的受累基因位于16号染色体末端16p13.3人珠蛋白基因(
HBA1
和
HBA2
)。该基因缺失或点突变可使α珠蛋白基因下调而抑制α珠蛋白链产生。目前发现的基因突变主要是结构基因的部分或全部缺失或
HBA1
和
HBA2
基因点突变,全球发现约65种不同的缺失,在中国人群中已鉴定出5种α
0
地中海贫血基因型(2个α基因都缺失),它们是:--
SEA
、--
THAI
、--
FIL
、--
HW
和--
11.1
;3种α
+
地中海贫血基因(1个α基因缺失),他们是:-α
3.7
、α
4.2
和-α
2.7
。--
SEA
是我国南方最常见的基因类型,其他常见突变依次为:-α
3.7
、-α
4.2
、α
CS
α和α
QS
α,这5种基因约占我国人群突变的90%。非缺失型的α地中海贫血由点突变、核苷酸插入等导致,很多突变影响mRNA进程、mRNA转录和α珠蛋白稳定性等,目前已发现约72种不同的突变。其中最常见的非缺失型变异为 α
IVSI
(-5nt)、多腺苷酸点突变
 。
。
3.临床表现
(1)静止型(α - /αα):
静止型α地中海贫血患者可无任何临床症状和体征,其父母一方有α地中海贫血。
(2)标准型(α - /α - 或--/αα):
标准型亦称α地中海贫血特征、轻型α地中海贫血。患者的临床症状轻,存在轻度贫血;其父母一方有α地中海贫血。
(3)Hb H病(--/α - ):
亦称中间型α地中海贫血。患者的临床表现多样,年幼时多无症状,约半数患者在20岁以后发病。多数患者病情较轻,主要表现为轻、中度的贫血和长期慢性溶血导致的肝脾大;重者则伴有黄疸;少数可伴骨骼轻微改变,不影响生长发育,因此无地中海贫血外貌。妊娠、感染、服用磺胺类或氧化剂药物时可诱发其溶血加重,某些严重者的表现与纯合子型β地中海贫血类似。往往患者的父母双方都有α地中海贫血。
(4)Hb Bart's胎儿水肿综合征(--/--):
一般患有Hb Bart胎儿水肿综合征的胎儿在妊娠30~40周时即可发生宫内死亡,或在早产或出生后数小时内死亡。患病胎儿皮肤苍白、全身水肿和各浆膜腔积液,伴或不伴有黄疸、皮肤出血、肝脾大,胎盘大而脆,脐带水肿明显。其父母均为--/αα型地中海贫血。
4.α地中海贫血的血液学筛查
(1)血常规筛查为首选方法,α地中海贫血红细胞重要特征是小红细胞和低色素,参考标准为:MCV<80fl;MCH<26pg。
(2)血红蛋白电泳技术,应用醋酸纤维素膜或琼脂糖作为电泳固相支持物,可对电泳分离出的不同血红蛋白进行定量分析。
(3)毛细管电泳技术有分析速度快,灵敏度高,分辨率高,样本用量和试剂消耗少,自动化程度高等特点。
(4)高效液相色谱(HPLC)可分析 Hb A2,国内将Hb A2<2.5%作为α地中海贫血的阳性指标。国外因人群不同,与国内的血常规筛查标准和血红蛋白标准存在差异。
5.α地中海贫血的基因诊断
α地中海贫血基因有两个富含GC、高同源性的α 1 和α 2 基因,可以使用高保真、热稳定性好的特异引物和特异反应条件进行扩增。经过多年的研究发展,目前,检测α地中海贫血突变类型的技术也不断的得到了提高,主要有下列8种技术可以选择:①Southern blotting印迹杂交技术,操作复杂,有放射性污染,但准确;②寡核苷酸(ASO)探针检测法,灵敏度高,须严格控制杂交条件,成本高;③突变特异性扩增系统法(ARMS)又称等位基因特异性扩增,是一种检测已知突变的方法,可鉴别非缺失型Hb H病基因型,该法简便、快速、经济、准确;④PCR-单链构象多态性检测(PCR-SSCP),该法复杂,多用于科研单位检测未知突变;⑤跨越断裂点PCR法(gap-PCR),普通PCR技术的缺点是只能诊断缺失纯合子而不能区分正常和杂合子样品,且容易产生假阳性结果,优点是可直接用于-- SEA 基因携带者的分子筛查,快速简便、经济准确;⑥DNA测序分析技术,可以鉴定未知突变,是基因检测的金标准方法;⑦反向点杂交法(RDB),其准确性仅次于DNA测序,为采用最广泛的诊断方法之一;⑧基因芯片技术,可将α、β地中海贫血基因诊断集中在一张芯片上完成,适合用于大面积普查。
6.治疗
α地中海贫血静止型和标准型患者临床无须治疗,注意膳食平衡,避免感染,必要时补充叶酸。重型患者偶有通过异基因造血干细胞移植获得成功的报道。由于目前对重型α地中海贫血总体尚无有效的治疗方法,因此,加强婚姻及遗传咨询、婚前及产前筛查,对夫妇双方为α地中海贫血基因携带者的妊娠妇女,在妊娠早期进行产前基因诊断,防止重型α地中海贫血儿出生以及减少杂合子胎儿出生,是目前最有效的预防措施。
(二)β珠蛋白生成障碍性贫血
亦称β地中海贫血(β-thalassemia),是指β链合成部分受抑(β+基因)或完全抑制(β 0 基因)而引起的一组血红蛋白病。
1.流行病学
全球大约有1.5%的人携带β地中海贫血基因,并且每年出生的患有纯合子或双重杂合子基因型的β地中海贫血新生儿至少有20万。在20世纪80年代,对我国20个省、市、自治区的60万人进行的血红蛋白病的调查显示,我国β地中海贫血的患病率约为0.67%。
2.分子机制
β地中海贫血的发生主要是由于珠蛋白基因突变或缺失,基因突变类型至少有186种,主要为点突变。比较常见的点突变类型为CD41~42、IVS Ⅱ 654、CD17、TATA-28、CD71~72、TATA-29等。上述珠蛋白肽链的基因发生突变,导致正常β珠蛋白肽链合成减少或不合成或产生异常肽链,从而使α珠蛋白肽链相对过剩形成包涵体沉积于红细胞膜上,使红细胞变形能力降低,易被破坏导致溶血性贫血。
3.临床表现
(1)重型β地中海贫血(β-thalassemia major):
亦称Cooley贫血,一般为β + 、β 0 基因纯合子或复合杂合子状态。患儿出生时正常,生后半年起逐渐苍白,重度贫血,黄疸,肝脾大。生长发育迟缓、矮小、肌张力松弛、常有发热及胃肠道症状。因长期骨髓增生,骨质疏松,骨骼生长畸形,并可引起病理骨折。颅骨增厚,额部隆起,鼻梁凹陷,二眼距增宽,呈特殊面容。X线检查见颅骨板障增宽,皮质变薄,骨小梁条纹清晰,似短发直立状;由于髓外造血灶,可压迫脊髓,产生相应神经症状。常表现重度贫血,呈低色素小细胞性贫血,靶形红细胞多见,网织红细胞升高。血红蛋白电泳分析示Hb F达30%以上,甚至可达100%,Hb A多低于40%,红细胞渗透脆性明显减低。骨髓红系增生超过正常的20~40倍,引起骨髓腔扩张,骨骼畸形。体内含铁血黄素沉积引起心、肝、肾等重要脏器功能损害。诊断根据典型病史,临床表现尤以特殊面容、骨骼X线表现、重度低色素小细胞性溶血性贫血及Hb F增多等。患者亲生父母应为轻型β地中海贫血患者。如无适当输血治疗,患者往往于婴幼儿期死亡。
(2)中间型β地中海贫血(β-thalassemia intermedia):
本组患者临床表现贫血程度轻于重型地中海贫血,一般不需经常输血,血红蛋白可维持在60~70g/L以上。患者贫血、黄疸程度不一,脾脏轻至中度肿大,少数病例有轻度骨骼改变,生长阻滞,性发育迟,但性功能仍能成熟。患者常可生存至成年,甚至老年,本组患者包含多种不同遗传基础,如症状较轻的纯合子β地中海贫血,贫血与脾大明显的杂合子β地中海贫血,β和δβ珠蛋白生成障碍性贫血,β和α珠蛋白生成障碍性贫血的双重杂合子,或β地中海贫血与Hb E、S、C、Lepore等异常血红蛋白的双重杂合子。
(3)轻型β地中海贫血(β-thalassemia minor):
父母中至少有一人患同样疾病。临床可无症状或仅轻度贫血,血红蛋白在90~110g/L,偶有轻度脾大。血片中可见少量靶形红细胞。红细胞较小,血红蛋白含量较低。红细胞渗透脆性减低,本病特征性表现为Hb A 2 升高,约90%以上患者Hb A 2 占3.5%~7.0%(正常 Hb A 2 2%~3%),约50%的患者Hb F水平增高至1%~3%,极少数大于5%。由于临床无明显症状,多在普查时发现。本病需与Hb H、δβ珠蛋白生成障碍性贫血等鉴别,并需与缺铁性贫血鉴别,后者Hb A 2 正常,骨髓可染铁、血清铁及血清铁蛋白均减低,铁剂治疗有效。
4.β地中海贫血的血液学筛查
同α地中海贫血。
5.β地中海贫血的基因诊断技术
(1)针对β地中海贫血“已知突变”的基因诊断:
在DNA分析技术上,对于基因缺失型β地中海贫血,可采用Gap-PCR和多重连接探针扩增(MLPA)技术进行分析;对于基因点突变型β地中海贫血,则可采用RDB技术进行分析,也可采用变性高效液相色谱分析(DHPLC)或基于实时荧光PCR的探针熔解曲线分析法来进一步验证RDB技术分析的结果。但Southern印迹杂交技术和DNA测序技术仍然是诊断β地中海贫血基因大片段缺失和点突变的金标准。
(2)针对β地中海贫血“未知突变”的基因诊断:
“未知”是指β地中海贫血基因的突变性质不明。对用上述技术手段分析后仍然不能鉴定突变类型的具有明显β-地中海贫血表型特征的个体则需要采用一些其他技术,如DHPLC、变性梯度凝胶电泳(DGGE)、单链构象多态性(SSCP)和MLPA等进行“未知突变”的筛查,以便能将这些不确定的基因突变锁定在某个DNA片段内,然后再利用DNA测序技术来分析这个包含有突变基因的DNA片段,从而能大大提高DNA测序的效率。
(3)植入前遗传学诊断(preimplantation genetic diagnosis,PGD):
PDG是辅助生殖技术与遗传学诊断技术相结合而形成的一种孕前诊断技术,是在体外受精的基础上,应用分子生物学技术对活检得到的卵母细胞的极体或胚胎的1~2个卵裂球进行遗传学分析,以去除携带严重遗传性疾病的胚胎,选择正常胚胎植入宫内,从而获得健康的胎儿。将PGD技术应用于地中海贫血的检测,作为产前诊断的一种更早期的方法,为那些不愿接受终止妊娠的地中海贫血高危夫妇提供了可供选择的手段。
6.治疗
(1)一般治疗:防治感染,禁用氧化剂药物,补充叶酸。
(2)输血治疗:高量输血治疗仍是治疗β地中海贫血的一种主要方法。输血治疗的主要目的是提高Hb水平,通过反复输血可使Hb维持在90~105g/L以上。但本法容易导致含铁血黄素沉着症,因此,如果长期实施高量输血治疗则必须同时给予铁螯合剂治疗。
(3)祛铁治疗:去铁胺(DFO)是临床上应用最广、效果较好的一种祛铁剂。DFO在血浆中有两种成分,并且以1∶1的比例与铁离子结合形成大分子稳定的铁胺复合物,从而从粪便和尿中排出。由于DFO口服吸收较差,并且在血浆中的半衰期短,因此只能采用持续皮下或静脉输注。通过高量输血和正规祛铁治疗,可维持患儿正常生长发育,达到正常人的生活质量及寿命。去铁酮(DFP)有许多不良反应,例如关节疼痛、一过性丙氨酸氨基转移酶升高、较严重的不良反应有粒细胞缺乏症和粒细胞减少症等。地拉罗司是一种新型口服铁螯合剂,能够排泄肝脏中的铁,有效预防和治疗β地中海贫血患者心脏的铁沉积。
(4)γ珠蛋白基因诱导药物:常用有细胞毒药物羟基脲(HU),其机制可能是由于其细胞毒作用对红细胞系的成熟和分化产生了影响,间接在细胞水平上增加了Hb F的含量。应用HU长期治疗,部分重型β地中海贫血患儿的贫血可获得改善,Hb F水平提升,肺动脉高压也能得到有效预防。DNA甲基转移酶(DNMT)抑制剂代表药物主要有5-氮胞苷(5-Aza)和地西他滨,可通过抑制DNMT而使γ-珠蛋白基因的启动子低甲基化而促进其表达。组蛋白脱乙酰基酶(HDAC)抑制剂是通过抑制HDAC来促进组蛋白H3、H4,特别是γ基因启动子区域内的组蛋白发生乙酰化。雷帕霉素靶点(TOR)抑制剂能诱导正常人及β地中海贫血患者红系前体细胞γ珠蛋白基因的表达。可以诱导Hb F生成的细胞因子有干细胞因子(SCF)、红细胞生成素(EPO)和转化生长因子(TGF)等。
(5)基因治疗:基因导入,将正常人的β珠蛋白基因导入β地中海贫血患者的骨髓细胞中,再回输给患者。基因 改造,用低分子量的核酸U1 snRNA、U2 snRNA或者U7snRNA对 β654 、 β705 等突变基因的非正常剪接位点进行反义封闭。
(6)造血干细胞移植是治愈重型β地中海贫血血患者的唯一手段。全相合同胞骨髓移植的无病生存率已提高到80%~87%。目前,非亲缘移植治疗效果已经达到同胞供者移植的疗效水平,非亲缘外周干细胞较移植的植入率更高、无病存活率更高。
三、不稳定血红蛋白病
不稳定血红蛋白病(unstable hemoglobinopathy,UHb)是由于α或β珠蛋白肽链与血红素的紧密结合的氨基酸发生替代或缺失,损害肽链的立体结构或减弱与血红素的结合力,形成分子结构不稳定的异常血红蛋白。UHb易受氧化而丢失血红素,在红细胞内聚集沉淀形成变性珠蛋白小体(Heinz小体),使红细胞膜变僵硬,易被脾破坏,导致溶血性贫血。以往曾称为先天性变性珠蛋白小体贫血。本病属常染色体显性遗传,但不少患者无家族史。迄今已发现200种以上不稳定血红蛋白,种类虽多但总发病率较低。绝大多数是由β链变异引起。各种不稳定血红蛋白的临床表现轻重不一,按其贫血程度可分为四组:①重度溶血性贫血:常为β链氨基酸替代或缺失引起,1岁以内起病,表现为慢性溶血性贫血,中到重度贫血40~80g/L,网织红细胞常在20%以上,切脾无效;②中度溶血性贫血:儿童或青春期起病,脾大,发作性黄疸,血红蛋白60~100g/L,切脾可使贫血减轻或消失;③轻度贫血或无贫血:网织红细胞轻度增高,感染及服用氧化剂药物均可引起溶血急性发作,甚至发生溶血危象,较常见的不稳定血红蛋白病如HbZürich、Hb Köhn属此组;④无贫血及任何临床表现,但实验室检查可有不稳定血红蛋白检出,多数患者在溶血发作时尿液呈深褐色或黑色,为血红素与珠蛋白肽链解离后产生的二吡咯色素尿。同时伴有高铁血红蛋白血症的患者可出现发绀。热变性试验(新鲜溶血液,加热50℃2小时后,如沉淀血红蛋白>5%,提示UHb存在)及异丙醇试验(新鲜溶血液加入异丙醇37℃10~15分钟,UHb可产生绒毛状沉淀)是诊断本病简便、敏感并具有一定特异性的试验。血红蛋白电泳对不稳定血红蛋白检出率不高,仅Hb Köhn、Hb-Sydney、HbZürich等少数不稳定血红蛋白可与Hb A分开而被检出。本病应与G6PD缺陷及其他血红蛋白病鉴别。对本病患者应注意预防感染和避免服用磺胺类及其他氧化型药物。急性溶血时应积极对症治疗。妊娠期贫血可加重。脾切除可使红细胞寿命延长,溶血减轻,对中度贫血患者效果较好,但对重型患者无效。
四、高铁血红蛋白血症及硫血红蛋白血症
正常人血红蛋白含二价铁(Fe 2+ ),与氧结合为氧化血红蛋白,如血红蛋白中Fe 2+ 被氧化为三价铁(Fe 3+ )时,称为高铁血红蛋白(Met Hb)。Met Hb氧亲和力高,使氧不能释放到组织,体内Met Hb>10%时,称高铁血红蛋白血症(methemoglobinemia)。
1.获得性高铁血红蛋白血症(acquired methemoglobinemia)
多种药物或有毒化学物质(如亚硝酸盐类、利多卡因、苯胺衍生物、磺胺类等)引起血红素铁氧化;临床症状严重度取决于Met Hb含量、发病速度及患者心、肺和造血系统对缺氧的代偿能力。Met Hb>15%时,常有脑缺血症状;达40%时可产生心悸、乏力、呼吸困难等缺氧表现;>60%时可出现虚脱、出汗、昏迷甚至死亡。慢性型多表现发绀,缺乏全身症状。不能用心、肺疾病解释的发绀,且吸氧无效者,应考虑本病。患者血液加入几滴10%氰化钾后,产生鲜红色氰化高铁血红蛋白,可与硫化血红蛋白及其他Met Hb鉴别。急性中毒性高铁血红蛋白血症是一种严重 的医疗急症。轻症患者(Met Hb 20%~30%)不需特殊处理,停服有关药物或脱离化学物质接触24~72小时后,Met Hb自行降至正常;Met Hb>40%,应给亚甲蓝1~2mg/kg加入25%葡萄糖液缓慢静注,对伴有G6PD缺乏的患者无效,且会诱发溶血,亚甲蓝应避免使用过量。维持治疗可口服亚甲蓝(60mg,3~4次/日)或维生素C(300~600mg/d)。西咪替丁可以减轻因氨苯砜引起的高铁血红蛋白血症。
2.先天性高铁血红蛋白血症(congenital methemoglobinemia)
遗传性细胞色素b5还原酶(b5R)缺陷(酶活力<20%)使Met Hb还原能力减低,常染色体隐性遗传,纯合子酶活力极低,血Met Hb可达50%~60%,患者生后即有发绀,但寿命正常。本病分单纯红细胞型及全身型(占10%~15%),前者血Met Hb 8%~40%,静脉血呈巧克力色。后者除高铁血红蛋白血症外,尚伴进行性脑病和智力低下,多数夭折。治疗使用维生素C(300~600mg/d,分3~4次口服),亚甲蓝静脉注射可暂时纠正高铁血红蛋白血症,但不宜长期应用。
3.血红蛋白M病(hemoglobin M disease)
珠蛋白基因突变,如血红素邻近的组氨酸被酪氨酸替代,使铁稳定在三价状态,已知多种Hb M(如 Boston、Iwate、Hyde Park和Saskatoon)均可引起高铁血红蛋白血症,本病患者用亚甲蓝及维生素C治疗无效,目前尚缺乏有效的治疗。
4.硫血红蛋白血症(sulfhemoglobinemia)
正常人血红蛋白中硫血红蛋白约占0~2%,高于正常为硫血红蛋白血症,常伴发于高铁血红蛋白血症,接触硝基甲苯、乙酰苯胺、农用杀虫剂代森锌等,均可引起本病,硫血红蛋白形成后不能逆转,含硫血红蛋白红细胞寿命及渗透性均正常,临床表现主要为缺氧发绀,有时可有轻度溶血,与高铁血红蛋白血症不易区分,亚甲蓝及维生素C治疗无效。
五、血红蛋白S病
血红蛋白S病(hemoglobin S disease)亦称镰状细胞病(sickle cell disease)。血红蛋白S(Hb S)是β珠蛋白链第6位谷氨酸被缬氨酸替代所致的异常血红蛋白
 )。由于分子表面电荷改变,在氧张力和pH减低时,高浓度的脱氧Hb S发生分子间的相互作用,在红细胞内聚合成液晶,其溶解度比氧合Hb S低五倍。脱氧 Hb S分子连接呈细丝状多聚体,细丝缠成中空螺旋形细长条,使红细胞由正常双凹盘状变形如镰刀状(镰变)。镰变红细胞膜僵硬,无法通过微循环,而引起局部缺氧。血黏稠度增加,小血管淤滞栓塞。当血红蛋白与氧结合时,分子间的相互作用消失,红细胞镰状化可迅速恢复正常。如红细胞膜明显受损,红细胞失钾失水,可导致镰状细胞不可回逆,引起微血管阻塞范围扩大成为大面积梗死,酸中毒或红细胞内2,3-二磷酸甘油酸增高,红细胞氧亲和力降低,增加脱氧血红素形成,进一步加重红细胞镰变。上述引起红细胞镰变的多聚体的形成和稳定均需β珠蛋白链的参与。Hb A含β链易参与Hb S多聚体的形成。Hb F不含β链,可抑制Hb S的多聚体化,故不含Hb F的红细胞易镰变。肾髓质的高渗环境中能引起局部红细胞镰变及肾乳头梗死形成。本病主要见于非洲与美洲的非洲后裔,前者Hb S杂合子约占20%,美洲的非洲后裔中约占8%~10%。我国已有报道,患者亲代系非洲黑种人。血红蛋白S病可分下列三种主要类型。
)。由于分子表面电荷改变,在氧张力和pH减低时,高浓度的脱氧Hb S发生分子间的相互作用,在红细胞内聚合成液晶,其溶解度比氧合Hb S低五倍。脱氧 Hb S分子连接呈细丝状多聚体,细丝缠成中空螺旋形细长条,使红细胞由正常双凹盘状变形如镰刀状(镰变)。镰变红细胞膜僵硬,无法通过微循环,而引起局部缺氧。血黏稠度增加,小血管淤滞栓塞。当血红蛋白与氧结合时,分子间的相互作用消失,红细胞镰状化可迅速恢复正常。如红细胞膜明显受损,红细胞失钾失水,可导致镰状细胞不可回逆,引起微血管阻塞范围扩大成为大面积梗死,酸中毒或红细胞内2,3-二磷酸甘油酸增高,红细胞氧亲和力降低,增加脱氧血红素形成,进一步加重红细胞镰变。上述引起红细胞镰变的多聚体的形成和稳定均需β珠蛋白链的参与。Hb A含β链易参与Hb S多聚体的形成。Hb F不含β链,可抑制Hb S的多聚体化,故不含Hb F的红细胞易镰变。肾髓质的高渗环境中能引起局部红细胞镰变及肾乳头梗死形成。本病主要见于非洲与美洲的非洲后裔,前者Hb S杂合子约占20%,美洲的非洲后裔中约占8%~10%。我国已有报道,患者亲代系非洲黑种人。血红蛋白S病可分下列三种主要类型。
1.Hb S纯合子(镰状细胞病,SCD)患者红细胞内Hb S浓度高,对氧亲和力显著减低,氧解离曲线右移,加速氧的释放,使患者能耐受严重缺氧。患者生后3~4个月,当Hb F被Hb S替代时,即开始出现症状和体征如苍白、黄疸、肝脾大、发育较差。由镰状红细胞阻塞微循环而产生的症状可遍及全身,引起脏器功能障碍,表现为腹痛、气急、肾区痛、血尿。发作性剧烈腹痛常被误诊为急腹症。此外尚有手、足、关节骨骼肿痛及下肢溃疡等。患者躯干短小、四肢细长,性成熟延迟。可因造血旺盛而发生叶酸缺乏性贫血,也可因再生障碍危象(特别是微小病毒B19感染引起的红系造血抑制)而使贫血突然加重,并发感染而致早年死亡,贫血通常表现为正色素正细胞性,其稳态血红蛋白水平在50~110g/L。血涂片易见镰形红细胞,重亚硫酸钠镰变试验可见大量镰状红细胞生成。血红蛋白碱性条件下电 泳,Hb S位于Hb A与Hb A 2 间;酸性电泳时,Hb S位于Hb A的阳极端。纯合子Hb S在80%以上,Hb F2%~20%,Hb A 2 正常,Hb A缺如。
2.Hb AS(镰状细胞特征)患者为 Hb S与Hb A基因杂合子,从双亲分别继承了一个正常β基因及一个异常β基因(β 6 Glu→Val )。患者一般无临床症状,血常规可正常,但在缺氧状态时(如全身麻醉、高空作业、严重肺部疾病等),红细胞可发生镰变,出现脾、肾梗死症状,表现为左上腹疼痛,无痛性血尿等。血红蛋白电泳示Hb A约60%,Hb S 35%,Hb A 2 正常。患者常同时伴有叶酸缺乏。伴缺铁时Hb S比例减少。患者寿命一般不受影响。
3.Hb S与β珠蛋白生成障碍性贫血杂合子,Hb S与β + 或β 0 基因双杂合子,如患者为β 6谷→缬 与β 0 基因杂合子,则临床表现类似Hb S纯合子。由于无正常β链生成,血红蛋白电泳显示无Hb A,Hb S浓度较高,Hb A 2 轻度增多, Hb F 5%~10%。常在幼年起病,伴严重溶血性贫血、血管梗死,常早年死亡。
Hb F可以改善SCD的表型,通过调节Hb F来治疗SCD。羟基脲起始剂量可为15mg/kg,每天给药1次,逐步加量至35mg/kg。可补充叶酸,积极预防感染和缺氧。溶血发作时应供氧及补液。发生梗死危象时,除上述治疗外,尚可使用血管扩张药及抗凝剂。国际血液及骨髓移植中心对镰状细胞病患者进行异基因骨髓移植,5年总生存率达 97%。在本病多发人群及地区,应进行Hb S普查,开展遗传咨询,劝阻杂合子婚配。对高危家系妊娠妇女,应作好产前诊断,提倡优生优育。
六、血红蛋白C病
Hb C(
 )是多见于非洲黑种人。西非加纳等发病率可达14%~28%,美国非洲裔人中Hb C杂合子亦可达2%~3%,我国尚无报道。Hb C的氧亲和力较低,氧合Hb C或含Hb C红细胞在高渗介质时,Hb C易形成细胞内结晶,故含Hb C的红细胞较正常红细胞僵硬,易在血液循环中丢失部分细胞膜而形成小球形细胞。本病可分为以下三型。
)是多见于非洲黑种人。西非加纳等发病率可达14%~28%,美国非洲裔人中Hb C杂合子亦可达2%~3%,我国尚无报道。Hb C的氧亲和力较低,氧合Hb C或含Hb C红细胞在高渗介质时,Hb C易形成细胞内结晶,故含Hb C的红细胞较正常红细胞僵硬,易在血液循环中丢失部分细胞膜而形成小球形细胞。本病可分为以下三型。
1.血红蛋白C病(hemoglobin C disease)
即Hb C纯合子。患者从父母各继承一个异常β基因(β 6Glu→Lys ),临床表现轻中度溶血性贫血,常伴轻到重度脾大、胆石症,血片中见靶形及小球形红细胞增多,偶可见红细胞内有长形结晶。血红蛋白电泳显示Hb C可高达90%以上,Hb A缺如,Hb F增多。本病无根治疗法,切脾疗效不明显。患者寿命一般不受影响,应注意防治感染,补充叶酸,对症治疗。
2.Hb C特征
即Hb C与Hb A杂合子。患者无临床表现,血常规检查正常,可见靶形红细胞。血红蛋白电泳显 示Hb C约40%,Hb A 2 、Hb F正常,余为Hb A,本病不需治疗。
3.镰状红细胞血红蛋白C病
即Hb C与Hb S杂合子,患者症状介于纯合子Hb S与杂合子Hb S患者之间,常于儿童青春期出现轻中度溶血性贫血、脾大,可发生视网膜病变、血尿,妊娠期易有并发症。治疗原则参照“血红蛋白S病”。
七、血红蛋白D病
Hb D是主要见于印度、巴基斯坦和伊朗。我国内蒙、新疆、青海、河北及河南等省区有少数病例散发。血红蛋白D病(hemoglobin D disease)不是一种单一的异常血红蛋白,其特点是在碱性pH电泳时迁移率与Hb S相同,而在pH 6.2琼脂电泳时与Hb S分离,与Hb A共迁移。无溶解度异常,镰变试验阴性。现已发现有几种β或α链氨基酸顺序不同的Hb D,最多见的是Hb D Punjab(或 Hb DLos Angeles,
 )。纯合子Hb D-Punjab无症状,无血液学异常,复合杂合子有轻度小细胞贫血和轻微溶血,无脾大,血涂片中有靶形红细胞。杂合子多无症状。而Hb D-Los Angeles和Hb S共遗传导致严重的镰形红细胞病表型,与纯合子Hb S相同。Hb D-Los Angeles应和Hb S相鉴别,可以联合使用常规血红蛋白酸性电泳和碱性电泳方法鉴别,等电点聚焦、HPLC和毛细管电泳技术也容易做出鉴别。
)。纯合子Hb D-Punjab无症状,无血液学异常,复合杂合子有轻度小细胞贫血和轻微溶血,无脾大,血涂片中有靶形红细胞。杂合子多无症状。而Hb D-Los Angeles和Hb S共遗传导致严重的镰形红细胞病表型,与纯合子Hb S相同。Hb D-Los Angeles应和Hb S相鉴别,可以联合使用常规血红蛋白酸性电泳和碱性电泳方法鉴别,等电点聚焦、HPLC和毛细管电泳技术也容易做出鉴别。
八、血红蛋白E病
Hb E(
 )最常见于东南亚,也为我国人群中较常见的异常血红蛋白,遍布南北16个省区,以广西壮族自治区及广东、云南省多见。其氨基酸替代位置虽在α
1
β
1
接触面上,但因谷氨酸和赖氨酸理化性质相似,对血红蛋白分子的稳定性和功能影响不大。血红蛋白E病(hemoglobin E disease)属常染色体不完全显性遗传,Hb E与其他珠蛋白变异体共遗传在其流行地区也很常见,其中最重要的是Hb E-β地中海贫血综合征。纯合子Hb E个体没有症状,常在筛查或家系研究过程中被诊断。Hb E-β地中海贫血是组异质性很大的疾病,临床表现可由中间型地中海贫血样表型至严重依赖输血的重型地中海贫血。纯合子Hb E常无明显贫血,但存在低色素小红细胞症,血涂片可见靶形红细胞,红细胞渗透脆性降低,血红蛋白电泳显示Hb E高达90%以上,Hb F为5%~10%,Hb A则缺如;在碱性pH电泳时,Hb E与Hb A
2
泳速相同,但 Hb E浓 度高,Hb A
2
即使在病理情况下,亦罕有高于10%者,以此可鉴别。Hb E对氧化剂不稳定,异丙醇试验多呈阳性,热变性试验也轻度阳性,部分患者变性珠蛋白小体生成率增高。Hb E纯合子患者不需要任何治疗。严重的Hb E-β
0
地中海贫血治疗参见中重型地中海贫血,应长期输血将血红蛋白维持在接近100g/L,若患者出现脾亢应考虑切脾。长期使用羟基脲或其他Hb F诱导剂可以提高患者Hb F水平,并降低红细胞无效生成。造血干细胞移植也有成功根治的报道。
)最常见于东南亚,也为我国人群中较常见的异常血红蛋白,遍布南北16个省区,以广西壮族自治区及广东、云南省多见。其氨基酸替代位置虽在α
1
β
1
接触面上,但因谷氨酸和赖氨酸理化性质相似,对血红蛋白分子的稳定性和功能影响不大。血红蛋白E病(hemoglobin E disease)属常染色体不完全显性遗传,Hb E与其他珠蛋白变异体共遗传在其流行地区也很常见,其中最重要的是Hb E-β地中海贫血综合征。纯合子Hb E个体没有症状,常在筛查或家系研究过程中被诊断。Hb E-β地中海贫血是组异质性很大的疾病,临床表现可由中间型地中海贫血样表型至严重依赖输血的重型地中海贫血。纯合子Hb E常无明显贫血,但存在低色素小红细胞症,血涂片可见靶形红细胞,红细胞渗透脆性降低,血红蛋白电泳显示Hb E高达90%以上,Hb F为5%~10%,Hb A则缺如;在碱性pH电泳时,Hb E与Hb A
2
泳速相同,但 Hb E浓 度高,Hb A
2
即使在病理情况下,亦罕有高于10%者,以此可鉴别。Hb E对氧化剂不稳定,异丙醇试验多呈阳性,热变性试验也轻度阳性,部分患者变性珠蛋白小体生成率增高。Hb E纯合子患者不需要任何治疗。严重的Hb E-β
0
地中海贫血治疗参见中重型地中海贫血,应长期输血将血红蛋白维持在接近100g/L,若患者出现脾亢应考虑切脾。长期使用羟基脲或其他Hb F诱导剂可以提高患者Hb F水平,并降低红细胞无效生成。造血干细胞移植也有成功根治的报道。
主要参考文献
1.郝颖,徐志勇,金晴,等.一种新的β地中海贫血基因突变类型的分子诊断和产前基因诊断.中华血液学杂志,2011,32(4):245-248.
2.王辉林,郭华,熊礼宽.α地中海贫血研究进展.中国热带医学,2011,11(9):1158-1160.
3.黄艳环,陆泠羽,苏国生,等.β地中海贫血临床诊治研究进展.慢性病学杂志,2014,15(8):619-622.
4.Goldman L,Schafer AI.Goldman's Cecil Medicine.25th ed.Philadelphia:Elsevier Saunders,2016,1089-1104.
第九节
自身免疫性溶血性贫血
陈勤奋
一、概 述
【定义】
自身免疫性溶血性贫血(autoimmune hemolytic anemia,AIHA)是B淋巴细胞功能异常亢进,产生抗红细胞自身抗体,与自身红细胞膜表面的抗原结合,然后活化补体,激活巨噬细胞,使红细胞破坏加速;或是自身抗体促进补体与红细胞结合,使红细胞寿命缩短,从而引起获得性溶血性贫血的一组疾病。
【分类】
根据自身抗体与红细胞反应的最佳温度,AIHA可分为温抗体型和冷抗体型两大类。尚有温冷抗体混合型,自身抗体为IgG温抗体和冷凝集素同存。
(一)温抗体型
与红细胞的最适反应温度为37℃(范围35~40℃)的自身抗体称为温抗体,它又可分为温性不完全抗体及温性自身溶血素。温性不完全抗体约占所有自身抗体的68.9%,常为IgG,吸附在红细胞膜上,经巨噬细胞吞噬使红细胞变为球形,通过单核-巨噬细胞系统时被大量吞噬而破坏,为血管外溶血,溶血主要发生在脾脏。温抗体又按化学结构不同分为IgG、IgM、IgA三类,主要是IgG,极少数为IgM,单纯IgA很罕见;IgG温抗体又可分为IgG1、IgG2、IgG3和IgG4亚型,主要是IgG1和IgG3,IgG2和IgG4少见。直接抗球蛋白试验(direct antiglobulin test,DAT)的结果为IgG阳性或IgG+C3阳性。
(二)冷抗体型
与红细胞的最适反应温度为0~5℃的自身抗体称为冷抗体。主要为完全抗体IgM,容易凝集红细胞,可结合补体,称为冷凝集素,多见于冷凝集素病(cold agglutinin disease,CAD),也称为冷凝集素综合征(cold agglutinins syndrome,CAS),为血管内溶血。另有一种特殊冷抗体,称为冷热溶血素(Donath-Landsteiner抗体,D-L抗体),为7SIgG抗体,见于阵发性冷性血红蛋白尿症(paroxysmal cold hemoglobinuria,PCH)。常见的 DAT结果是IgG阴性C3阳性。
(三)混合型
温抗体与冷抗体各自识别红细胞膜上的抗原,反应温度在4~37℃,通常红细胞破坏更为严重。
根据病因的有无,AIHA还可分为原发性(原因不明性)和继发性两大类。由于诊断技术日趋完善,继发性病例的比例逐渐增高。见表16-2-10。
表16-2-10 AIHA的病因分类

【流行病学】
AIHA可在儿童(主要在5岁前)和成人发病,年发病率约为1/10万~3/10万,儿童患者男性多于女性,成人患者女性∶男性约为1.5~2.0。成人患者多数超过40岁,发病高峰年龄70岁,这可能与淋巴增殖性肿瘤在老年人群高发相关。绝大多数为散发病例,家族性病例罕见。
AIHA通常以单一的免疫性“血细胞减少”出现,但有时也会同时或先后伴有免疫性血小板减少(称为Evans综合征)或自身免疫性中性粒细胞减少。
在AIHA中,温抗体型占成人病例的70%~80%,儿童病例的90%;CAD约占AIHA的7.7%~25.0%,几乎都在50岁以上发病;PCH约占AIHA的1.7%~5.1%,可发生在所有的年龄组,但以儿童为常见,男女比约为2.1∶1,无种族差异;混合型少见,发病以50岁以上居多,男女之比为1∶1.5。
二、温抗体型自身免疫性溶血性贫血
【发病机制】
AIHA的发病是一个复杂的多步骤的过程,不仅涉及自身抗体,还涉及免疫系统的各种效应,包括补体系统、巨噬细胞以及B、T淋巴细胞。引起溶血的机制已部分阐明,主要包括抗体依赖、细胞介导的细胞毒以及补体依赖的细胞毒。
(一)红细胞抗体
红细胞自身抗体的产生源于B淋巴细胞对自身红细胞抗原产生的免疫反应,这种免疫反应一方面是由于自身免疫耐受的破坏,另一方面是由于免疫调节的异常。树突状细胞和T、B淋巴细胞在自身免疫耐受过程中起重要作用,而Th1/Th2细胞的平衡以及调节性T细胞(Treg)则在免疫调节中起重要作用。Treg缺乏与AIHA发病密切相关。
1.自身免疫耐受状态的破坏
免疫耐受机制包括中枢耐受和周围耐受,前者是指骨髓和胸腺自然淘汰对自身抗原特异的T和B淋巴细胞,后者是指自身抗原特异性的B细胞由于缺乏T细胞提供的第二信号而不能增殖和分化。免疫耐受遭破坏后,免疫系统就会对自身细胞或组织发动体液或细胞免疫介导的攻击,造成自身免疫性疾病。
2.免疫调节的异常
CD4 + Th细胞分为Th1和Th2,前体为Th0细胞,可在不同的微环境诱导下向Th1或Th2细胞分化,两类细胞动态平衡,维持细胞与体液免疫呈稳定状态。AIHA患者Th1/Th2细胞失衡,Th2细胞功能异常亢进,产生过量IL-4、IL-6和IL-10等B细胞调控因子,导致体液免疫亢进。CD4 + 、CD25 + 的Treg细胞占CD4 + 细胞的10%~15%,是一种调节性免疫抑制细胞,Treg细胞功能异常,机体易患自身免疫性疾病。
3.免疫监视功能的异常
某些疾病如淋巴增殖性疾病会造成免疫监视或识别功能的紊乱,不能识别自身抗原而产生自身抗体。
4.遗传素质
HLA-Ⅰ类抗原中A1、A3和B8频率升高,HLA-Ⅱ类抗原中HLA-DQ6与溶血程度呈负相关。
(二)红细胞的破坏
温抗体型AIHA的自身抗体多为不完全抗体,致敏红细胞在通过单核-巨噬细胞系统时被巨噬细胞识别,巨噬细胞的Fc-γ受体(Fc-γ)与IgG的Fc片段结合,完整吞噬致敏红细胞或部分红细胞膜使其变成球形红细胞,最后在脾脏破坏,发生血管外溶血。Fc-γ有三种类型:Fc-γⅠ、Fc-γⅡ和 Fc-γⅢ。Fc-γⅠ与IgG 1 及IgG 3 有高度亲和力,然后通过Fc-γⅡ和Fc-γⅢ对吸附有自身抗体的红细胞发生吞噬作用。IgG 1 与Fc-γⅢ结合后的主要反应为吞噬作用,而IgG 3 与Fc-γⅢ结合后则为细胞毒溶解,最后都在脾内破坏。因此IgG 3 对致敏红细胞的破坏远较其他亚型严重,IgG 4 几乎无反应。温抗体的Fc还有C1q的结合位点,可激活补体C1,但只能到达C3阶段,不能形成C5~C9膜攻击复合物,不造成血管内溶血。
温抗体型AIHA的溶血速度不仅与红细胞表面自身抗体的数量、亲和力相关,也与巨噬细胞的数量和活动度相关。贫血程度则与红细胞的破坏速度和骨髓红系代偿增生的能力相关。
【临床表现】
贫血为主要症状,以黄疸为首发表现者较少见。原发性AIHA通常呈慢性过程,但也有数天内突发严重贫血和黄疸者。临床症状依患者年龄、贫血程度、原发或继发、有无其他基础疾病如冠心病等而不同。在稳定代偿阶段,红细胞可接近正常范围。约1/3的患者有苍白及黄疸;半数以上有轻至中度脾大,质硬不痛;1/3有中等肝大,不痛。淋巴结多不肿大。急性发病多发生于小儿,特别是伴有感染者,偶见于成年,起病急骤,有寒战、高热、腰背痛、呕吐和腹泻,可有休克及头痛、烦躁以致昏迷等神经系统表现。继发性患者常伴有原发疾病的临床表现,患者会首先因原发疾病而就诊。血栓栓塞性疾病发病率升高,尤其以抗磷脂抗体阳性者为甚,发生血栓时应注意筛查抗磷脂抗体。妊娠可促使温抗体型AIHA加重或诱发首次发作,但绝大多数症状较轻,如治疗及时,胎儿总体预后良好。
在感染和叶酸相对缺乏时可发生危象,有的患者以危象为首发表现而就诊。危象有两种:一种为溶血危象,特点有:①贫血突然加重,黄疸加深;②血管外溶血,尿色呈浓茶样,血管内溶血则有血红蛋白尿,尿色呈葡萄酒色或酱油色;③网织红细胞明显增高;④脾大;⑤一般白细胞和血小板数正常;⑥骨髓为增生性贫血象。
另一种为再生障碍危象,特点有:①贫血突然加重,但黄疸不加深;②网织红细胞减低,甚而阙如;③全血细胞减少,如为纯红再障危象则白细胞和血小板数正常;④骨髓象增生减低,类似再生障碍性贫血,如为纯红再障危象,则仅红系减少或阙如,粒系和巨核系正常。
【实验室检查】
(一)血常规
为正细胞、正色素性贫血,血片上可见多数球形红细胞,1/3的患者有数量不等的幼红细胞,偶见吞噬红细胞现象。网织红细胞多增高,极个别可达50%。由于网织红细胞增高,MCV增大;由于网织红细胞和球形红细胞的不同形态,RDW也增大。半数以上的患者白细胞数正常,急性溶血阶段白细胞增多,甚至有类白血病反应,但也有并发白细胞减少和粒细胞减少。血小板数正常或增多,有些患者在病程中发生免疫性血小板减少性紫癜,称为Evans综合征。
(二)骨髓象
特征性地表现为幼红细胞增生性骨髓象,粒/红比例倒置,偶见红细胞轻度巨幼样变。发生再障危象时骨髓呈增生低下,全血细胞及网织红细胞减少。
(三)红细胞破坏增加的生化检查指标
血清总胆红素升高,以非结合胆红素为主,乳酸脱氢酶升高,急性溶血时结合珠蛋白降低,但这些指标均为非特异性。
(四)抗球蛋白试验(antiglobulin test,Coombs test)
是诊断AIHA的经典实验室检查,有直接(DAT)和间接(indirect antiglobulin test,IAT)两种。DAT采用多价抗血清测定吸附在红细胞膜上的不完全性抗体(IgG、IgA、IgM)和补体(C3),称为广谱DAT试验;IAT检测患者血清中游离抗体或补体,诊断价值不如DAT重要,但对诊断药物诱发的免疫性溶血性贫血及同种抗体介导的溶血有价值。广谱DAT阳性,还需用特异单价抗血清检测以分亚型。亚型有三型:①抗IgG和抗C3型占67%,红细胞破坏最甚,贫血最重;②单独抗IgG阳性占20%,溶血中等;③单独抗C3型占13%,溶血最轻。抗IgA和抗IgM型均罕见。
DAT阳性必须每个红细胞上至少有300~400个IgG分子或60~115个补体C3分子,低于此数时常规DAT阴性。因此,抗体量不够或低亲和力抗体被洗脱时可能出现假阴性。若用更敏感的检测方法如生物素亲和系统-抗球蛋白试验(BAS-Coombs)、亲和素生物素化酶复合物酶联免疫分析(ABC-ELISA)、抗IgG消耗、免疫荧光法、流式细胞术检测红细胞结合IgG等可能检出抗红细胞抗体。真正DAT阴性的AIHA极少。
DAT阳性不一定有溶血性贫血。在正常献血者和普通住院患者中,可检出1/100~1/15 000不等的DAT阳性而无临床溶血表现,称DAT假阳性,主要是由于红细胞表面吸附有非特异性、低亲和力的IgG。通过洗脱红细胞上的抗体并检测其结合到正常红细胞上的能力可以区别DAT阳性的真假。假阳性时,洗脱的抗体不能结合到正常红细胞上,真阳性时,抗体可以结合到正常红细胞上。其他假阳性的原因有:①标本中有血块微粒;②用硅胶管采血;③抽静脉血混有低离子强度溶液;④高免疫球蛋白血症;⑤某些药物如头孢菌素可致血浆蛋白非特异性吸附于红细胞表面。极少数情况下并不是假阳性,而是AIHA进展的一个预兆。DAT阴性IAT阳性可能系同种免疫抗体所致,与输血或妊娠相关,而不是自身免疫所致。
【诊断与鉴别诊断】
诊断可按下列4个步骤进行:①贫血是否为溶血性贫血;②溶血是免疫性的还是非免疫性的;③如为AIHA,进一步鉴定抗体的类型;④确定AIHA是原发还是继发。凡近4个月内无输血或特殊药物服用史的溶血性贫血患者,如DAT阳性,结合临床表现和实验室检查,可确立诊断。如DAT阴性,但临床表现较符合,糖皮质激素或切脾治疗有效,除外其他溶血性贫血特别是遗传性球形红细胞增多症,可诊断为DAT阴性的AIHA。诊断原发性AIHA一定要排除继发病因,仔细询问药物史和相关疾病史可提供线索。
AIHA外周血常有球形红细胞增多,要与遗传性球形红细胞增多症鉴别。有的AIHA红细胞可有PNH样缺陷,使Ham试验阳性,CD55和CD59表达减少,需与PNH鉴别。其鉴别点在于AIHA时:①CD55和CD59缺陷仅见于自身抗体致敏的红细胞而骨髓有核红细胞表达正常;②粒细胞表达CD55和CD59正常或轻度异常;③缓解后CD55和CD59表达恢复正常。
【治疗】
(一)病因治疗
积极寻找病因,治疗原发病。药物性AIHA,停用药物即能纠正溶血。若由淋巴瘤引起,经化疗缓解后溶血可纠正;若由感染所致,控制感染后即可缓解甚至治愈。
(二)支持治疗与输血
无论哪种类型的AIHA,在溶血活动期补充叶酸(5~10mg/d)可预防因红系增生所致的叶酸耗竭,避免治疗失败。输血适用于溶血危象,或极重度贫血短期内可能危及生命,或有冠心病、心衰等基础疾病者。少数患者因自身抗体所致的自发性红细胞凝集可造成血型鉴定及交叉配血困难,应与输血科沟通,输血科可用温盐水洗涤患者红细胞,将红细胞表面吸附的抗体洗涤后再做定型试验;或提供交叉配血“最小不相容”的血液输注。应避免大量、快速输血,因为输入的红细胞可能被自身抗体破坏,也可能因既往输血或妊娠产生的异体抗体而加重溶血。温抗体所针对的血型抗原主要是Rh抗原系统,配血时应注意避开。
(三)一线治疗
糖皮质激素,是治疗AIHA首选和主要的药物,其作用机制可能为:①抑制抗体的产生;②降低抗体对红细胞膜抗原的亲和力;③减少巨噬细胞的Fc-γ和C3受体数量。糖皮质激素仅对温抗体型疗效较好,约80%以上的患者可获得早期完全或部分缓解,但停药后多数患者复发,长期应用不良反应较显著。起始剂量为泼尼松1~1.5mg/(kg·d),如治疗有效,待血红蛋白接近正常后每周减量一次,每次日量减5~10mg,直至30mg/d。以后每隔1~2周从日量中减去5mg,直至10~15mg/d,维持2~3个月,然后再每隔2周在日量中减去2.5mg。如出现复发,则须回复至先前最后一次有效剂量,至获得疗效为止。如足量治疗3周病情无改善,则视为治疗无效。无效者或维持量超过15mg/d者,应考虑其他疗法。与利妥昔单抗联用可增加有效率和有效持续时间。
(四)二线治疗
1.达那唑
用于辅助治疗IgG型AIHA,400~800mg/d,对糖皮质激素依赖、泼尼松剂量>15mg/d方能维持缓解的患者可减少糖皮质激素剂量,但对于复发和Evans综合征患者疗效较差,对IgM型AIHA无效。有男性化副作用和长期应用潜在的肝脏毒性。
2.利妥昔单抗
是一种与B淋巴细胞表面CD20抗原具有高度亲和力的嵌合体单抗,可迅速清除循环中和淋巴组织中的B淋巴细胞,通常剂量为375mg/m 2 ,每周1次,4~8次,用于对糖皮质激素、切脾和免疫抑制剂无效的难治性患者。需注意可能引起过敏反应,并可增加感染的危险性。其他单抗如阿仑珠单抗(抗CD52)和那他珠单抗(natalizumab)用于难治性、输血依赖病例已有少量文献报道。
3.脾切除(splenectomy)
脾既是温抗体型AIHA致敏红细胞破坏的主要场所,又是产生抗体的器官。脾切除适应证是:①糖皮质激素治疗无效;②糖皮质激素维持量>10mg/d;③不能耐受糖皮质激素治疗或有糖皮质激素应用禁忌证。总有效率为60%~75%。一般不适用于冷抗体型AIHA,因冷性抗体往往是依赖于补体的完全抗体,可直接在血液循环中引起溶血,凝集的红细胞有时在肝内破坏。最主要的危险是术后感染。推荐腹腔镜脾切除术。有切脾禁忌者可行脾区放射治疗。
(五)免疫抑制剂
包括细胞毒类和非细胞毒类药物。最常使用的细胞毒类免疫抑制剂有硫唑嘌呤、环磷酰胺和甲氨蝶呤等。硫唑嘌呤50~200mg/d,环磷酰胺50~150mg/d,开始3个月与糖皮质激素合用,以后糖皮质激素减量以致停用,再单用免疫抑制剂6个月。需密切观察不良反应,尤其是骨髓抑制。非细胞毒类免疫抑制剂有环孢素、吗替麦考酚酯等。免疫抑制剂适应证是:①糖皮质激素或切脾治疗不能缓解者;②脾切除有禁忌证者;③泼尼松维持量>10~20mg/d者。
(六)其他
包括静脉丙种球蛋白、血浆置换。静脉丙种球蛋白广泛用于治疗ITP,同样可用于治疗AIHA,400mg/(kg·d),连用5天,或2g/(kg·d),连用2天,但疗效不如ITP。血浆置换疗效有限,因温抗体IgG型AIHA是血管外溶血,大部分抗体在红细胞表面而非血浆里。但在严重病例,血浆置换可能挽救生命。
【预后】
继发性AIHA的预后取决于原发疾病的控制程度。原发性AIHA通过糖皮质激素或(和)脾切除治疗,75%的患者贫血能被纠正。少数患者溶血证据可消失,但多数患者DAT持续阳性,可有轻度溶血复发,需间断用糖皮质激素治疗。
【Evans综合征】
AIHA同时或相继发生免疫性血小板减少时称为Evans综合征(Evans syndrome),可出现于部分成人和儿童的急性AIHA中,国内统计约占AIHA的17.8%~23.0%,女性多于男性(3.3∶1)。少数病例还可伴有显著的粒细胞减少,可诊断为免疫性全血细胞减少,这部分病例的自身抗体直接针对红细胞、白细胞和血小板。多数病例原因不明,少数可继发于慢性淋巴增殖性疾病和SLE、类风湿关节炎、皮肌炎、桥本甲状腺炎、甲亢等自身免疫性疾病,肿瘤、感染、药物和妊娠等亦可诱发。自身抗体以IgG+C3型居多,占70%~100%,C3型17.6%~25.5%,IgG 型11.8%~14.9%,也有冷抗体或温冷抗体混合型、Coombs试验阴性的Evans综合征。病程慢性迁延,容易复发。通常糖皮质激素用于控制急性发作,有些持续性血细胞减少的患者需要延长糖皮质激素疗程或更积极的治疗。脾切除可能获益,儿童患者切脾感染风险增高且易复发。
三、冷凝集素病
【发病机制】
(一)冷凝集素
正常血清含有低滴度无功能的冷凝集素,滴度在1∶10以上时不能被检测出来,慢性CAD的冷凝集素滴度可超过1∶10 5 。冷凝集素在体外与红细胞的作用温幅很宽,但几乎所有的病例最易在4℃时凝集红细胞而在37℃时迅速分离。冷凝集素与红细胞抗原的亲和力弱,表现为温度依赖,反应可逆。多数冷凝集素主要针对红细胞表面的i抗原和I抗原,少数针对Pr抗原。由于把i抗原转化为I抗原的酶在出生后才活化,因此成人红细胞上都是I抗原而没有i抗原。支原体肺炎感染相关的冷凝集素具有抗-I特异性,传染性单核细胞增多症相关的冷凝集素具有抗-i特异性,慢性CAD冷凝集素几乎都是单克隆IgM。抗-I抗体的重链段由 VH4-34 基因编码,约有10%的正常B细胞有 VH4-34 基因片段,这可能就是正常血清中低滴度冷凝集素的来源。少数冷凝集素不针对I/i抗原系统而与Pr抗原直接结合,抗Pr冷凝集素的基因位于轻链可变区κⅣ。
(二)红细胞的破坏
在CAD中,红细胞破坏的基础是IgM抗体固定补体的能力。与IgG抗体不同,IgM分子有两个C1q结合位点,一个IgM分子足以结合C1,启动补体瀑布。正常红细胞上有补体抑制物,能抑制溶血作用。受寒的血液提供冷凝集素与红细胞结合,启动红细胞表面C3b和C4b的产生。进入温暖的内脏循环后,红细胞释放冷凝集素,但保留了C3片段。补体C1和免疫球蛋白重链Fc段结合后,经过经典的补体活化途径,活化后的C3插入红细胞膜,引起膜渗透性的不稳定,造成血红蛋白漏出。该过程的效力依赖于红细胞表面冷凝集素的数量、冷凝集素激活补体途径的能力以及热波幅。热波幅比冷凝集素效价更具有临床意义,仅在非常低温时有活性的冷凝集素所导致的损害很轻,而在接近37℃时有活性的冷凝集素导致的溶血则很严重。
【临床表现】
在寒冷环境下耳廓、鼻尖、手指发绀,进入温暖环境即消失。主要症状为贫血和黄疸,寒冷天气可加重。由于血清中存在C3b抑制物,贫血通常不严重,无须治疗可稳定数月。
高亲和力的冷凝集素遇冷可使红细胞在毛细血管循环中大量凝集,手足发绀,发生血管内溶血,出现血红蛋白尿,罕见情况下出现急性肾衰竭。手足发绀与Raynaud现象不同,没有苍白、反应性充血和局部坏疽,也不是冷球蛋白血症的血管炎。脾大不常见,除非与B细胞肿瘤相关。
【实验室检查】
1.室温时红细胞有自凝现象,加热后凝集现象消失常提示CAD的可能性。血液凝集使红细胞形成钱串状,可能干扰自动血细胞计数仪。
2.慢性轻度至中度贫血,外周血中无红细胞畸形,球形红细胞不明显,网织红细胞计数与贫血的程度成比例。可有轻度的高胆红素血症,反复发作者可有含铁血黄素尿。
3.冷凝集素试验阳性,4℃时效价高达1∶1000,甚至1∶16 000,30℃时在白蛋白或生理盐水内凝集效价仍高者有诊断意义。
4.如果冷凝集素有宽温幅,则DAT阳性,几乎全为C3型。
5.如有B细胞淋巴瘤,可在血清中检出单克隆IgM冷凝集素。
【诊断与鉴别诊断】
冷凝集素试验阳性,效价较高(>1∶40),结合临床表现和其他实验室检查可诊断为CAD。DAT为C3阳性而IgG阴性。尚需和PNH、PCH和冷球蛋白血症进行鉴别,进一步做冷热溶血试验和冷球蛋白试验以分别除外PCH和冷球蛋白血症。红细胞自凝集现象明显,应与红细胞钱串形成区别,自凝集现象于加热至≥37℃时可消失。
【治疗】
原发慢性CAD往往处于稳定状态,治疗主要是避免寒冷。继发于感染的急性CAD以保暖及支持疗法最为重要,溶血程度重时需及时输注预加温至接近37℃的红细胞悬液。继发于肺炎链球菌感染应使用抗生素,继发于病毒(EBV)感染的严重CAD可考虑用短程糖皮质激素以缩短病程。切脾无效。在慢性CAD和B细胞淋巴瘤患者,治疗主要针对肿瘤,抑制单克隆IgM的合成,降低抗体滴度,减轻溶血程度。可用苯丁酸氮芥和环磷酰胺,但疗效不确切。对烷化剂耐药的原发性CAD可考虑使用氟达拉滨,虽然已知氟达拉滨会引起AIHA。利妥昔单抗(375mg/m 2 每周1次静脉输注,4~8次)在某些病例有效。因IgM的分子量大,且37℃时绝大部分脱落在血浆里,血浆置换治疗CAD较温抗体型AIHA更有效,需用5%的白蛋白做置换液,以避免血浆中的补体加剧溶血。
【预后】
CAD的预后较温抗体型AIHA为好,多数患者能耐受轻度贫血,对劳动力影响较少,能长期生存,仅少数严重病例死于贫血或输血反应。
四、阵发性冷性血红蛋白尿症
【发病机制】
红细胞表面产生的Donath-Landsteiner抗体(D-L抗体),低温时可直接与红细胞膜上的P抗原结合并激活补体C1q,37℃时D-L抗体与红细胞分离,但C1q仍结合在红细胞膜上并引发补体瀑布式反应,造成红细胞膜真性穿孔,导致血管内溶血。D-L抗体多为多克隆抗体。
【临床表现】
发作前有寒冷环境暴露史,即使仅有单侧肢体暴露,也可在返回到温暖环境时即引发急性血管内溶血,典型表现为寒战、高热(高达40℃)、全身无力、腰背疼痛、腹部不适和血红蛋白尿,荨麻疹常见,但急性肾衰竭罕见,贫血是严重血管内溶血的结果。发作为自限性,多数持续数小时,偶有持续几天者。
【实验室检查】
发作时贫血严重,进展较迅速,急性期有网织红细胞减少这个显著特征,但最终会出现网织红细胞增多。周围血液可见红细胞大小不一及畸形,并有球形红细胞、红细胞碎片、嗜碱点彩及多染性红细胞,甚至幼红细胞。初始可有短暂的白细胞减少,随后有白细胞增多。血小板数通常正常或增高。红细胞渗透脆性增加常伴有球形红细胞增多。反复发作后有含铁血黄素尿。冷热溶血试验(D-L试验)阳性,试验操作应体现寒冷的患者血液加热至37℃时在试管里发生溶血,常常需要加入正常血清作为补体来源来引发溶血反应。DAT为C3阳性。
【诊断与鉴别诊断】
除典型临床表现外,D-L试验阳性可诊断本病,但该试验相对不敏感。诊断尚需排除CAD、PNH、行军性血红蛋白尿、肌红蛋白尿。详细询问病史与尿色改变的关系及有关特征性检查,不难鉴别。
【治疗】
以治疗原发病、保暖及支持治疗为主。通常2~3天后即症状消失,数周或数月内可自行缓解。切脾无效。
【预后】
PCH虽在急性发作时症状严重,但一般不致成为慢性严重贫血或死亡的原因。儿童严重贫血可导致猝死。D-L抗体可持续多年。
五、温冷抗体混合型自身免疫性溶血性贫血
血清中既有温抗体(主要为IgG、G3,极少数为IgM、IgA)又有冷凝集素的AIHA,称为温冷抗体混合型(mixed warm-and cold-antibody)AIHA。温抗体型AIHA约35%有低效价冷凝集素(4℃时≤1∶64),在20℃时可凝集红细胞,在30℃时失去活性,对细胞破坏无明显作用。少数AIHA除温抗体外还有冷活性IgM抗体,4℃时效价高,30~37℃时仍有活性,为高温幅的冷凝集素,此为真正兼有温冷抗体的AIHA。临床表现复杂,主要为严重贫血、黄疸,多数患者有肝脾大,可有Raynaud现象、肢端发绀或血红蛋白尿,也可表现为Evans综合征。受冷不一定加重病情。DATIgG和C3多阳性,冷凝集素试验阳性(常<1∶64),有宽温幅特征(4~37℃均有活性),D-L抗体阴性。红细胞自凝集现象强阳性。糖皮质激素治疗反应差,宜加用环孢素、环磷酰胺、长春新碱、硫唑嘌呤等免疫抑制剂。血浆置换仅作为急救辅助治疗。脾切除疗效不肯定。
主要参考文献
1.张之南,沈悌.血液病诊断及疗效标准.第3版.北京:科学出版社,2007:68-75.
2.Goldman L,Schafer AI.Goldman's Cecil Medicine.25th ed.Philadelphia:Elsevier Saunders,2016:1073-1078.
3.Kaushansky K,Lichtman MA,Kipps TJ,et al.Williams Hematology.8th ed.New York:McGraw-Hill,2010,957-985.
4.Greer JP,Arber DA,Glader B,et al.Wintrobe's clinical hematology.13th ed.Philadelphia:Lippincott Williams&Wilkins,2014,746-765.
5.Lechner K,Jäger U.How I treat autoimmune hemolytic anemias in adults.Blood,2010,116(11):1831-1838.
6.Berentsen S.How I manage cold agglutinin disease.Br J Haematol,2011,153(3):309-317.
7.Bass GF,Tuscano ET,Tuscano JM.Diagnosis and classification of autoimmune hemolytic anemia.Autoimmun Rev,2014,13(4-5):560-564.
8.Zanella A,Barcellini W.Treatment of autoimmune hemolytic anemias.Haematological,2014,99(10):1547-1554.
第十节
母婴血型不合溶血病
周文浩
母婴血型不合溶血病又称新生儿溶血病(Hemolytic Disease of the Newborn,HDN),是指母婴血型不合引起的同族免疫性溶血,仅见于胎儿和新生儿期,是新生儿期黄疸和贫血的重要原因。胎儿红细胞所具有的血型抗原恰为母亲所缺少时,胎儿红细胞通过胎盘进入母体循环,可使母体产生对胎儿红细胞抗原相应的抗体,此抗体(IgG)又经胎盘循环抵胎儿循环,作用于胎儿红细胞使其致敏并导致溶血,这是新生儿溶血病发病的基本原理。在我国以ABO血型系统母婴不合引起溶血者最为常见,其次为Rh血型不合,其他如Kell、Duffy、Kidd、MNSs等抗原性较弱的血型系统不合引起的新生儿溶血病极为少见。
【发病机制】
胎儿红细胞在妊娠30多天时即具有ABO和Rh系统抗原。母胎间的胎盘屏障并不完善,妊娠早期即可发生母亲至胎儿及胎儿至母亲的输血。妊娠3个月时在母体血液中可检测到胎儿红细胞。大多数妊娠妇女血中的胎儿血量仅0.1~3.0ml。但若反复多次小量胎儿血液进入母体,则可使母体致敏。
1.Rh血型不合溶血病(Rh blood type incompatible hemolytic disease)
Rh血型系统共有5种抗原,即D、C、c和E、e,这些抗原被位于1号染色体短臂上的RHCE和RHD基因所编码,RHCE基因编码Rh抗原C/c和E/e蛋白,RHD基因编码Rh抗原D蛋白,不存在D抗原的隐性等位基因“d”抗原。D抗原的免疫源性最强,是引起新生儿溶血病的主要原因之一,含有该抗原者称为Rh阳性血型,不含该抗原者称为Rh阴性血型。
Rh溶血病在第一胎发病率很低(1%左右),因为尽管在整个孕期都可有少量胎儿红细胞进入母体循环,可引起母亲致敏的明显量的胎母输血发生在妊娠末期或临产时,而初次免疫反应的产生需要2~6个月,且为IgM抗体不能通过胎盘,故第一胎常处于原发性免疫反应的潜伏阶段而不发病。一旦母亲已经致敏,再次妊娠暴露于RhD抗原时,就会产生IgG记忆反应,仅需数日就可出现以IgG为主的抗体,能够通过胎盘使胎儿红细胞致敏而发生溶血。由于Rh系统的抗体只能由人类红细胞引起,第一胎发生的Rh溶血病提示母亲以前曾经接触过Rh阳性红细胞。妊娠高血压、剖宫产、胎盘早期剥离、异位妊娠、臀位产、前置胎盘等产科因素及羊膜腔穿刺、经腹部穿刺绒毛活检、流产等可增加胎儿血液进入母体的机会,故可增加发生Rh溶血的危险性。
Rh阴性的频率在种族中有很大差异,白种人群中约占15%,我国某些少数民族(如维吾尔族)中也占5%以上,但在汉族人群中仅占0.34%。RhD溶血病主要发生在Rh阴性母亲和Rh阳性的胎儿,但Rh溶血病也可发生于母婴均为Rh阳性时,其中以抗E较为多见,我国汉族人群中无E抗原者几占半数,其他如抗C或抗e、抗c也可引起新生儿溶血病。
2.ABO血型不合溶血病(ABO blood type incompatible hemolytic disease)
ABO血型系统的基因位于9号染色体(9q34),基因ABO及H控制着A、B抗原的形成。除红细胞外,ABO血型物质还广泛存在于自然界。母亲产生ABO免疫抗体的途径有:①O型妇女曾经接受过不同血型红细胞抗原的刺激,如误输ABO血型不合的血液,或在妊娠、分娩、流产时,不同血型的胎儿血液进入母体;②O型妇女曾经接受过非特异性免疫刺激,如预防接种、某些食物或细菌的刺激等;③O型妊娠妇女曾经接受过含有A、B血型物质的胎儿组织、体液的刺激。因此,ABO溶血病可发生于第1胎。
在ABO血型不合溶血病中,由于O型血孕母所产生的抗A或抗B免疫抗体为IgG抗体,可通过胎盘进入胎儿循环而引起胎儿红细胞凝集溶解。而A或B型血孕母产生的抗B或抗AIgG抗体滴度较低。因此ABO血型不合所致的新生儿溶血病多见于O型母亲所生的A或B型胎儿(新生儿),而罕见A型母亲所生的B型胎儿(新生儿)或B型母亲所生的A型胎儿(新生儿)。A型或B型母亲所生的B型或A型新生儿发生的溶血病不足ABO溶血病的5%。
ABO血型不合溶血病的发病机制与Rh溶血病相似,即母亲的抗A或抗B抗体进入胎儿循环,与胎儿红细胞表面的A或B抗原反应。但是,ABO血型抗原的免疫原性与Rh血型系统的肽类抗原不同,其为碳水化合物抗原,不依赖T细胞,产生抗体主要为IgM,不伴有记忆反应,也不随反复接触抗原而增加抗体亲和力。A型和B型血者天然的抗B和抗A抗体主要为不能通过胎盘的IgM,然而,由于细菌、病毒或食物中AB抗原的持续刺激,同种免疫抗体可存在于O型血的个体内,包括能够通过胎盘的IgG抗体,因此,ABO血型不合溶血病仅限于O型母亲和A型或B型的胎儿,且常发生于第一胎(40%~50%)。
与Rh溶血病相比较,ABO溶血病相对较轻,这是因为A和B抗原也存在于红细胞外的许多组织中,通过胎盘的抗A或抗B IgG仅小部分与红细胞结合,其余都被其他组织的AB血型物质所吸收。
【临床表现】
新生儿溶血病的临床表现可以轻重不一,Rh溶血病和ABO溶血病的临床特点比较见表16-2-11。一般而言,在ABO溶血病中,新生儿A型血型者较B型者多见,但B型者病情较A型者重。
表16-2-11 Rh溶血病和ABO溶血病的比较

1.轻度溶血
此型以ABO溶血病最为常见,受累患儿仅有轻度溶血,几乎没有贫血(脐血Hb>14g/dl),轻度髙胆红素血症(脐血胆红素<4mg/dl)。除早期光疗外,新生儿一般不需要治疗。
2.中度溶血
发生在少数ABO溶血病和多数Rh溶血病患儿中。表现为中度贫血(脐血Hb<14g/dl)和脐血胆红素增加(>4mg/dl)。黄疸常在生后24小时内出现,具有出现早、上升快等特点,若不治疗,可发生胆红素脑病。肝脾明显肿大,外周血有核红细胞增多。血清胆红素以未结合胆红素为主,但也有少数患儿在病程恢复期结合胆红素明显升高,出现“胆汁瘀积”。由于血型抗体持续存在,患儿可在2~6周发生明显贫血(Hb<80g/L),称晚期贫血。晚期贫血也可因换入的成人红细胞氧亲和力较低和氧运增加而抑制了红细胞的生成所致。
3.重度溶血
主要见于Rh溶血病,患儿可死于宫内,或出生时胎儿水肿,表现为全身水肿、苍白、皮肤淤斑、胸腔积液、腹水、心力衰竭、肝脾大和呼吸窘迫。胎儿水肿原因除重度贫血致高心排量心力衰竭外,还与肝功能障碍致低蛋白血症和组织缺氧致毛细血管通透性增高有关。黄疸可在生后迅速出现并进行性加深。此型病情极重,如不及时治疗,常于生后不久死亡。除晚期贫血外,其他血液学并发症还包括血小板减少症、凝血障碍、白细胞减少等。
【诊断及干预】
1.出生前诊断及干预
目的为减轻病情,治疗贫血。抗体筛查应在第一次产前检查或孕24~28周时通过间接抗人球蛋白试验(Coombs试验)测定。当检测到任何抗体时都应进行免疫分型及抗原的特异性测定。如果该抗体为IgG,并已知是对抗红细胞的某一抗原时,即应开始严密的产前监护。
(1)定期监测抗体滴度和胎儿基因分型:
随访时间通常为妊娠中期每2~4周1次,妊娠晚期每1~2周1次。提示胎儿有可能受损而需要进行有创试验(如羊膜腔穿刺或直接胎儿血标本采样)的最低抗体滴度随抗原的特异性而不同,常用的RhD溶血病的临界滴度为1∶16或1∶32。孕早期通过羊水细胞或绒毛测定胎儿RhD分型可准确识别处于溶血风险的胎儿,提供母亲选择终止妊娠的机会,胎儿Rh阴性也可尽早使家属放心和避免进一步操作。胎儿超声是评估母婴血型不合妊娠的基本手段,有助于证实胎儿疾病的严重程度和早期发现胎儿水肿,超声引导在胎儿脐血采样和宫内输血时也必不可少。超声评估应包括胎儿解剖学、胎盘形态学和羊水量。多普勒血流速度监测的目的是检测胎儿因贫血而致血液黏度降低时的血流动力学代偿性变化。
(2)出生前治疗
1)胎儿宫内输血(intrauterine transfusion,IUT):
对于在宫内严重受累(血细胞比容≤30%)的胎龄<32周和肺功能不成熟的胎儿,可在B超引导下胎儿脐血管穿刺直接血管内红细胞输注。输入血应为与母亲血清不凝的抗原阴性的浓缩红细胞,容量按输注后血细胞比容达到40%计算,可重复。
2)孕母注射静脉用免疫球蛋白(IVIG):
在孕28周前,胎儿受累较重但尚未发生胎儿水肿者,可给孕母注射IVIg400mg/(kg·d),疗程4~5天。间隔15~21天可以重复,直至分娩。IVIG可反馈抑制母体血型抗体的产生、阻止母体抗体经胎盘进入胎儿体内;IVIG也可与胎儿巨噬细胞上Fc受体结合,抑制血型抗体所致红细胞破坏。
3)提早分娩:
如果宫内输血操作困难或胎儿不能耐受,在35周之前计划早产也是一个可选择的方法。在计划分娩之前,应当从羊水评估胎儿肺的成熟性,并在产前48小时供给糖皮质激素促胎肺成熟。母亲分娩前1周开始口服苯巴比妥可诱导胎儿肝酶成熟,减少生后换血需要。
2.出生后诊断及处理
及时纠正贫血和黄疸,避免胆红素脑病的发生,可以及早发现。
(1)对疑有母婴血型不合溶血病的新生儿应进一步作下列实验室检查:
红细胞及血红蛋白下降(脐血<14g/dl),网织红细胞增高(>6%)、外周血有核红细胞增高(>10/100个白细胞)等均提示患儿可能存在溶血。但确诊本病的主要依据是血清特异性免疫抗体的检查。Rh血型不合者Coombs试验直接法阳性即可确诊,并可作释放试验以了解是哪种Rh血型抗体。将婴儿血清与各种标准红细胞作Coombs间接试验,阳性结果表明有血型抗体存在,并可根据与标准红细胞的凝集反应推论抗体类型。新生儿红细胞上存在的A或B抗原位点相应较少,因此ABO溶血病直接Coombs试验反应微弱,需作改良法Coombs试验、抗体释放试验及游离抗体三项试验。其中改良法直接Coombs试验和(或)抗体释放试验阳性均表明新生儿的红细胞已致敏基本可以确诊,以释放试验阳性率较高。若仅游离抗体阳性只能表明新生儿体内有抗体,并不一定致敏,故不能作为确诊依据。
(2)新生儿治疗:
新生儿溶血病的防治目的:①预防严重贫血和低氧所致的宫内或出生后不久的死亡;②避免由于高胆红素血症所致的胆红素脑病。
1)产房复苏及胎儿水肿的处理:
儿科医生应该参与分娩和心肺复苏。脐血标本立即测定新生儿血型、抗体滴度、血红蛋白和胆红素浓度。如果出生时即有胎儿水肿、严重贫血、高心排量心衰或休克的体征,应保持有效通气、抽腹水或胸腔积液和尽快换血。危重者可先用浓缩红细胞小量输血或部分交换输血以纠正贫血,然后再以正常红细胞比容的2倍血容量进行换血。
2)大剂量IVIG在新生儿溶血病中的应用:
出生后一旦明确诊断为新生儿溶血病,可尽早静脉滴注1剂IVIg500mg~1g/kg,于2小时内滴入。因IVIG可有效地阻断新生儿巨噬细胞Fc受体,抑制溶血过程,使胆红素产生减少,从而减少换血。但因缺乏大样本IVIG治疗新生儿溶血病安全和有效的临床试验证据,目前尚不推荐IVIG作为新生儿溶血病的常规治疗。
3)连续监测血清未结合胆红素和预防核黄疸:
对高胆红素血症者应采取积极措施(光疗或换血)降低血清胆红素和保持内环境稳定,以避免胆红素脑病的发生。未结合胆红素升高速率可作为判断溶血严重程度和是否需要换血的指标。常用的标准为:胆红素上升速率>0.5mg/(dl·h),或在出生后头2天每24小时上升>5mg/dl,或血清胆红素水平超过预期换血标准(足月儿为20mg/dl)。
4)纠正贫血:
早期贫血严重者往往血清胆红素很高而需换血。晚期贫血若程度不重可进行观察,但当小儿因贫血而心率加快、气急或体重不增时应适量输血。输血的血型应不具有可引起发病的血型抗原和抗体,可输注浓缩红细胞,每次10~20ml/kg。也有学者应用促红细胞生成素(EPO)治疗晚期贫血取得效果,但尚缺乏大样本的临床证据。
5)胆汁淤积的监测:
由于溶血和宫内输血造成的铁过载是引起胆汁淤积症的重要因素,此外,胆色素过载导致胆汁小管淤滞、贫血缺氧导致肝坏死、髓外造血的压力导致肝内毛细胆管受压以及感染、代谢性疾病、全肠外营养等均系易患因素。虽然目前临床资料显示溶血所致的胆汁淤积症可在生后1周至3个月自发缓解,但是在此期间直接胆红素和肝脏酶学的监测是非常必要的。
主要参考文献
1.Gottstein R,Cooke RW.Systematic review of intravenous immunoglobulin in haemolytic disease of the newborn.Arch Dis Child Fetal Neonatal Ed,2003,88(1):F6-10.
2.Murray NA,Roberts IA.Haemolytic disease of the newborn.Arch Dis Child Fetal Neonatal Ed,2007,92(2):F83-88.
3.Roberts IA.The changing face of haemolytic disease of the newborn.Early Hum Dev,2008,84(8):515-523.
4.Anstee DJ.The relationship between blood groups and disease.Blood,2010,115(23):4635-4643.
第十一节
药物、感染及理化因素引起的溶血性贫血
袁 燕
一、药物引起的溶血性贫血
(一)药物诱发的免疫性溶血性贫血(drug-induced immune hemolytic anemia,DIIHA)
少见,发病率为0.1/10万,据报道约有127种药物认为和DIIHA相关,其中42%为抗微生物药,16%为非甾体抗炎药,13%为抗肿瘤药,6%为降压药。20世纪80年代引起DIIHA的主要药物为α-甲基多巴和大剂量青霉素。近年来随着上述药物临床应用的减少,80%的病例是由第二、三代头孢菌素和β内酰胺酶抑制剂引起,特别是头孢替坦、头孢曲松和哌拉西林。嘌呤类似物氟达拉滨、2-氯脱氧腺苷和脱氧助间型霉素引起的DIIHA也日益增多,慢性淋巴细胞白血病应用氟达拉滨治疗发生率可达5%~22%。
药物分子量小,称“半抗原”,具有抗原性,和大分子载体蛋白(红细胞膜蛋白)结合,可诱发免疫反应产生抗体,该抗体可只针对药物和药物代谢物,或部分药物部分载体蛋白,或只针对载体蛋白。前两种涉及的抗体是药物依赖型,在间接抗人球蛋白试验(IAT)体系中需要有药物存在才能获得阳性结果;第三种涉及的是药物非依赖型抗体,IAT试验无须药物存在,后者实际上是红细胞真性自身抗体;某些药物可能存在多种机制(混合型抗体)。此外,有些药物(主要是头孢菌素)结合红细胞膜后可非特异性吸附血浆中的IgG,出现直接抗人球蛋白试验(DAT)阳性,但不出现溶血。头孢替坦、头孢曲松和哌拉西林诱发的DIIHA都是药物依赖型抗体,氟达拉滨则是引起真性自身抗体最常见的药物。
DIIHA的临床表现无特异性。可以是药物暴露后立刻引起急性血管内溶血致严重溶血性贫血,也可以是用药数月后引起轻度血管内溶血。确定药物暴露史有时也有一定困难,否认有暴露史的患者不能排除以前进食过污染有抗生素的食品。由于绝大多数DIIHA病例DAT试验为阳性,因此极易误诊为原发性温抗体自身免疫性溶血性贫血(WAIHA)。如有明确的药物暴露史,停药后溶血缓解即可诊断DIIHA,具有真性自身抗体的DIIHA和WAIHA无区别。从体外要正确诊断是何种药物诱发也有一定难度,当然IAT、药物洗脱试验有一定帮助,切勿用可疑药物做激发试验。
一旦疑及DIIHA,应立即停药,贫血严重具有药物依赖型抗体者应输血,必要时血浆置换。凡属真性自身抗体引起者可使用糖皮质激素或其他治疗WAIHA的方法。
(二)药物诱发非免疫性溶血性贫血
某些药物(主要是氧化剂),可使血红蛋白氧化变性,形成高铁血红蛋白和Heinz小体,或产生自由基和过氧化物过多,形成变性血红蛋白,均可导致红细胞破坏。特别是当红细胞内还原系统有缺陷或血红蛋白异常时更易发生。
二、感染引起的溶血性贫血
感染性疾病常出现红细胞寿命缩短,尤其是伴有G6PD缺陷症、脾大和微血管病的患者。引起溶血性贫血的微生物种类繁多,有曲霉菌、炭疽杆菌、巴贝虫、巴尔通体、空肠弯曲菌、梭状芽孢杆菌、肺炎链球菌、大肠埃希杆菌、嗜血流感杆菌、脑膜炎球菌、沙门菌、志贺菌属、链球菌、小肠结肠炎耶尔森菌、EB病毒、柯萨奇病毒、巨细胞病毒、肝炎病毒、单纯疱疹病毒、人类免疫缺陷病毒、流感病毒、水痘病毒、风疹病毒、麻疹病毒、疟原虫、杜氏利什曼原虫、钩端螺旋体、结核分枝杆菌、肺炎支原体、弓形虫属、布氏锥虫、霍乱弧菌等。感染引起溶血性贫血的机制有:病原体直接侵犯红细胞引起红细胞破坏;产生溶血毒素;产生抗体或免疫复合物导致溶血;增强巨噬细胞识别红细胞和引发噬血细胞综合征等。
疟疾是引起溶血性贫血最常见的病因。以恶性疟和间日疟引起的病例数最多。间日疟原虫仅侵犯网织红细胞,而恶性疟原虫可以侵犯各期红细胞,所以后者导致的溶血更为严重。
巴尔通体不侵入红细胞内,仅吸附于红细胞外表面。一种130kD的巴尔通体蛋白可以使红细胞膜内陷及出现凹痕沟槽,导致溶血。血涂片Giemsa染色可发现长1~3μm,宽0.25~0.30μm的紫红色小体。
巴贝虫是一种红细胞内原虫,可通过蜱虫叮咬或者输血传播,病原体可在红细胞内繁殖继而导致红细胞裂解。薄血涂片Giemsa染色在红细胞内发现虫体即可诊断,也可以通过血清学或PCR技术检测来诊断。
梭状芽孢杆菌脓毒血症可致严重急性溶血。这种细菌可分泌α溶血素,其中卵磷脂酶可水解红细胞表面的卵磷脂蛋白,使红细胞形成球形,易在脾脏内破坏。
三、物理、化学因素与生物毒素所致的溶血性贫血
(一)物理因素引起的溶血性贫血
物理因素引起的溶血性贫血主要源于机械性损伤和热损伤。
红细胞通过部分阻塞的血管或经过血管的异常表面时,会因受到剪切力的作用使细胞破裂导致溶血,可在患者外周血片见到红细胞碎片。见于心脏瓣膜修复术(包括人工瓣膜、同种瓣膜、异种瓣膜和自身瓣膜成形术),腱索断裂及心内补片修复术,瓣膜疾病(特别是主动脉瓣狭窄)和主动脉缩窄。也可见于行军性血红蛋白尿症等。微血管病性溶血性贫血也是属于机械性损伤所致(见本章第十二节“其他类型的贫血”)。
严重烧伤后可出现急性血管内溶血(多在24~48小时内),高温(>47℃)可使红细胞发生不可逆的形态和功能异常,其严重程度与烧伤面积、温度高低和持续时间相关。
(二)化学因素引起的溶血性贫血
可引起溶血性贫血的非氧化剂类化学物包括砷化氢、铜、铅、高浓度氧等。砷化氢吸入后95%~99%以非挥发形式与血红蛋白结合,2~24小时即可出现中毒表现,病死率可高达20%。可以选择换血疗法去除含砷的红细胞。误服硫酸铜和长期使用含铜管路进行血透导致铜蓄积可发生溶血性贫血。肝豆状核变性(Wilson病)因血铜水平增高也可导致溶血。大量铜可抑制葡萄糖-6-磷酸脱氢酶的活性,从而导致红细胞破坏。血浆置换可用于治疗肝豆状核变性患者的急性溶血。铅等重金属中毒时,可导致红细胞5'端核苷酸酶(P5'N)活性获得性损害,从而损伤红细胞。高浓度的氧气可使红细胞脂质成分异常过氧化导致溶血。氧化剂类化学物致溶血性贫血见本章第八节。
(三)生物毒素导致的溶血性贫血
毒蛇种类很多,不同种毒蛇其蛇毒的组成成分亦不同,大多数蛇毒含有多种活性成分。蛇咬伤后引起溶血性贫血并不多,远比神经症状和消耗性凝血病为少,溶血可以是急性爆发的,也可以延至数小时或数天后发作。蛇毒中多种成分可以导致溶血,其中磷脂酶A2,对红细胞膜有直接毒性,有些蛇毒中含有活性酶可以激活补体引起溶血。棕色隐士蛛咬伤后部分患者可出现血管内溶血,病程通常1周内自限,但偶有严重病例引发肾衰竭及死亡。某些毒蜂蜇伤后可在12~24小时后出现迟发型反应,包括溶血、DIC和肝肾功能损害等。蜂毒中含有蜂毒肽和磷脂酶A2,可协同损伤红细胞膜导致溶血。
主要参考文献
1.Garratty G.Immune hemolytic anemia associated with drug therapy.Blood Reviews,2010,24(4-5):143-150.
2.Goldman L,Schafer AI.Goldman-Cecil Medincine.25th ed.Philadelphia:Elsevier Saunders,2016,1078.
3.Means RT Jr,Glader B.Wintrobe's clinical hematology.13th ed.Philadelphia:Lippincott Williams&Wilkins,2013,809-821.
第十二节
其他类型的贫血
王小钦
一、急性失血后贫血
因外伤或疾病致血管破裂,或凝血、止血障碍等使大量血液在短期内丢失,可致急性失血后贫血(acute post-hemorrhagic anemia),主要影响血容量。
急性大量失血的主要病理生理改变是血容量急骤减少,动脉血压降低。早期代偿机制是通过心血管动力学的调整及肾上腺素能的刺激作用,使心率加快、心输血量增加,循环血量重新分配,以保证重要脏器组织以及对缺氧敏感器官如心、肺、肝、脑组织的血液供应。并通过红细胞内合成更多的2,3-二磷酸甘油酸及血红蛋白氧离曲线右移,使组织氧释放增加。该期的主要临床表现是血容量不足。2~3天后血容量的恢复主要依靠水、电解质和白蛋白从血管外被动员入血,使血浆容量扩增,血液被稀释,另一方面血红蛋白浓度和血细胞比容不断下降,出现贫血。急性失血引起组织缺氧,可刺激肾脏产生EPO,促进骨髓幼红细胞增生,骨髓的代偿能力取决于骨髓造血功能、EPO的反应以及铁供应是否充沛。
贫血是正细胞正色素性。网织红细胞在急性失血后2~3天内开始升高,6~11天达高峰。急性失血后2~5小时白细胞也迅速增高,主要是中性粒细胞增多,核左移,可出现幼粒细胞。失血后1~2小时血小板开始升高,个别可达1000×10 9 /L。白细胞和血小板多在3~5天恢复正常,如果持续升高,必须排除潜在出血的可能。急性内出血,血液进入体腔和组织间隙,因红细胞破坏出现游离胆红素和乳酸脱氢酶升高,结合珠蛋白降低加上网织红细胞增多,酷似溶血性贫血,须注意鉴别。
治疗原则首先应当针对出血的原因立即设法止血,另一方面要采取紧急措施补充血容量,防止休克的发生。迅速输入生理盐水、血浆、右旋糖酐、白蛋白或羟乙基淀粉,并尽早输入全血或红细胞。对贫血本身一般不需特殊治疗,应在度过急性期后及早给予高蛋白质、富维生素的饮食,如果体内贮铁充分,造血功能正常,则红细胞在出血停止后4~6周恢复正常,血红蛋白的恢复要落后2周。原有慢性失血史或原先贮铁量不足者,出血停止病情稳定后可给予铁剂,以促进红细胞的生成和铁贮量的补充。
二、慢性肾病贫血
慢性肾病贫血(anemia of chronic kidney disease)是指肾衰竭所致的贫血。慢性肾衰如有感染或炎症的持续存在可致ACD;慢性失血可引起IDA;反复血透可引起巨幼细胞贫血;溶血尿毒综合征可致微血管病性溶血性贫血等。在rhEPO使用之前,98%慢性肾衰血透患者有贫血,由于rhEPO的使用,贫血的患病率已有显著降低。
其发病机制主要与肾产生EPO不足相关。此外,肾衰患者还能通过炎症因子(TNF-α、IL-6及IFN-γ等)使EPO产生减少,并通过hepcidin使铁吸收、转运和利用减少,幼红细胞凋亡增加,因此肾衰患者有EPO产生减少常同时伴功能性缺铁。尿毒症毒素可直接抑制红系生成,红细胞寿命缩短也是肾性贫血的原因之一。
贫血为正细胞正色素性,后期可以出现小细胞低色素性。贫血程度与肾衰程度相关,可达重度贫血。红细胞形态多数正常,网织红细胞数正常或轻度升高。白细胞数和血小板数通常正常。骨髓可呈轻度增生,如肾衰发展快,亦可呈红系增生减低。根据贫血、肾功能不全、EPO水平下降、排除其他原因的贫血可以诊断为肾性贫血。
肾性贫血治疗的指征为Hb<100g/L,治疗目标为Hb在110~120g/L,不超过130g/L。首选rhEPO治疗,初始剂量为50~100U/kg,每周3次,或10 000U每周1次,希望每月Hb上升10~20g/L,如2周内Hb上升超过20g/L,将剂量减少25%。如果治疗效果不佳,可以增加EPO剂量。血透患者应用EPO治疗时,补铁要及时,当转铁蛋白饱和度<30%,或血清铁蛋白<500μg/L就需要补铁,静脉补铁优于口服补铁,每3个月监测铁蛋白。推荐应用蔗糖铁,100mg每周1次,一个疗程的总剂量为1000mg。当转铁蛋白饱和度≥50%,或铁蛋白≥800μg/L时停止补铁。
EPO失效或存在EPO抵抗,要考虑是否缺铁、缺乏叶酸、透析不足、近期有感染或炎症、继发性甲旁亢、铝中毒、血管紧张素转换酶抑制剂的使用以及有EPO的抗体等因素。
三、慢性肝病贫血
慢性肝病贫血(anemia of chronic liver disease)系指慢性肝病患者伴有轻中度贫血,但须除外因肝病的并发症,如上消化道大量出血引起失血性贫血,酗酒致酒精性肝病所致铁粒幼细胞性贫血,因肝病营养障碍缺乏叶酸所致巨幼细胞贫血等。约75%的慢性肝病伴有贫血,机制不明。肝硬化的患者全身血容量常较正常高10%~15%,因稀血症加重了贫血的发生率和程度。
贫血为轻、中度。多为正细胞性,可以有轻度大红细胞性贫血,可见靶形红细胞、棘细胞。酒精性肝病伴溶血发作,同时具有黄疸、高脂血症,就称Zieve综合征。无特殊治疗,肝功能好转后贫血自然纠正。
四、内分泌疾病相关性贫血
许多内分泌激素参与红细胞生成的调节,包括甲状腺素、糖皮质激素、睾酮和生长激素。因此甲状腺功能减退症(hypothyroidism)、甲状腺功能亢进症(hyperthyroidism)、肾上腺皮质功能减退症(adrenocortical insufficiency)、垂体功能减退症(hypopituitarism)、甲状旁腺功能亢进症(hyperparathyroidism)等均可导致贫血,一般为轻、中度贫血。激素替代治疗可纠正贫血。
甲状腺功能减退时,EPO分泌减少,多数为正细胞正色素性贫血,如为小细胞低色素性贫血需考虑合并缺铁;如有显著大红细胞贫血,考虑是否合并维生素B 12 缺乏。甲减贫血合并恶性贫血的概率增加。单纯甲减贫血补充甲状腺激素,贫血纠正是逐步的,约需6个月Hb才能恢复正常。甲亢贫血常有小红细胞增多,其机制不详。甲亢治疗后,贫血及小红细胞增多均可纠正。
慢性肾上腺皮质功能减退症几乎所有病例均有贫血,但因有脱水而被掩盖,激素替代治疗后,因血浆容量扩充,初Hb反下降,以后才逐渐上升。3%~16%非结核性肾上腺皮质功能减退可见恶性贫血。垂体功能减退常有轻度贫血。
五、骨髓病性贫血
骨髓病性贫血(myelophthisic anemia)是骨髓被恶性肿瘤或其他异常细胞广泛浸润以及肉芽和(或)纤维组织大量增生,使骨髓结构破坏影响红系造血所致的贫血。由于血液骨髓屏障破坏及肝、脾和淋巴结的髓外造血,使幼稚细胞进入血流。其特征除贫血外周围血液出现幼粒、幼红细胞,故又称幼粒-幼红细胞性贫血(leuko-erythroblastic anemia)。
本症常见的病因是:①肿瘤组织浸润骨髓,见于造血系统肿瘤和非造血系统肿瘤,如乳腺癌、肺癌、前列腺癌、胃癌等。②骨髓纤维化,包括原发性和继发于慢性粒细胞白血病、癌症以及血管炎等。③肉芽肿性炎症,如结核和真菌感染。④代谢异常,类脂质沉积症如戈谢病和尼曼-皮克病及骨硬化症,后者系单核-巨噬系统遗传性缺陷,表现为破骨细胞功能减低。
贫血程度不一,呈正细胞正色素性。血片中可见多染性、嗜碱性点彩、异形及泪滴状红细胞,并出现幼粒细胞和幼红细胞,甚至巨核细胞裸核。白细胞和血小板高低不一。骨髓穿刺常有干抽现象,骨髓涂片和活检可发现原发疾病的表现,癌症转移时骨髓涂片可见成堆癌细胞。骨髓活检对骨髓纤维化或肉芽肿的诊断更有价值。
在诊断骨髓病性贫血时,必须注意周围血液出现幼粒和幼红细胞也见于大量失血、溶血、MDS及类白血病反应等,应加以鉴别。
治疗以针对原发病为主。对贫血本身可选用雄激素及EPO。
六、微血管病性溶血性贫血
微血管病性溶血性贫血(microangiopathic hemolytic anemia,MHA)又称红细胞破碎综合征,是一组继发性贫血综合征,主要是由于红细胞在血管内受机械性损伤破坏所致。可见于下列疾病:①血栓性血小板减少性紫癜(throm-botic thrombocytopenia purpura,TTP)、溶血性尿毒症综合征(hemolytic uremic syndrome,HUS)及弥散性血管内凝血(disseminated intravascular coagulation,DIC),是其临床表现的一部分。②心脏和大血管病变,特别是心脏人工瓣膜置换术后。目前人工瓣膜质量已显著提高,术后MHA出现率已很少见。③恶性肿瘤广泛播散,肿瘤细胞可以直接侵犯微血管,受累血管内皮细胞增生,管腔内有微小瘤栓,其中以胃癌广泛转移为最多见,其次为乳腺癌、肺癌、胰腺癌等。④化疗相关的MHA,这些患者绝大多数使用了化疗药物如丝裂霉素C,器官移植后也可发生,与预处理化疗药物损伤内皮细胞有关。⑤妊娠相关的MHA,妊娠期和产后均可发生,子痫和子痫前兆可以诱发,发生HELLP综合征(hemolysis,elevated liver enzymes and low platelets syndrome)。
诊断标准为:①有皮肤、黏膜不同程度的出血和黄疸、贫血、发热等表现;②外周血涂片中破碎红细胞3%以上;③骨髓红系增生明显活跃,网织红细胞增多;④血小板计数明显减少;⑤血浆游离血红蛋白增高,常>50mg/L;⑥间接胆红素增高;⑦结合珠蛋白降低;⑧血红蛋白尿,慢性者可有含铁血黄素尿。满足第①、②项标准,再有其他两项指标,即可诊断。
需与自身免疫性溶血性贫血、Evans综合征、阵发性睡眠性血红蛋白尿、DIC等鉴别。
治疗原则为积极治疗原发病,去除病因(例如心脏瓣膜的重新置换,肿瘤的控制,自身免疫性疾病的治疗等)。慢性溶血者如有缺铁表现,可给予铁剂和叶酸。严重贫血时可以输红细胞。血浆置换仅在少数病例有效。
七、先天性红细胞生成异常性贫血
先天性红细胞生成异常性贫血(congenital dyserythropoietic anemia,CDA)是一组罕见的遗传性贫血。主要特点是:①骨髓幼红细胞形态有病态表现如多核、核碎裂和巨幼样变(图16-2-17);②无效造血;③继发性铁负荷过多。
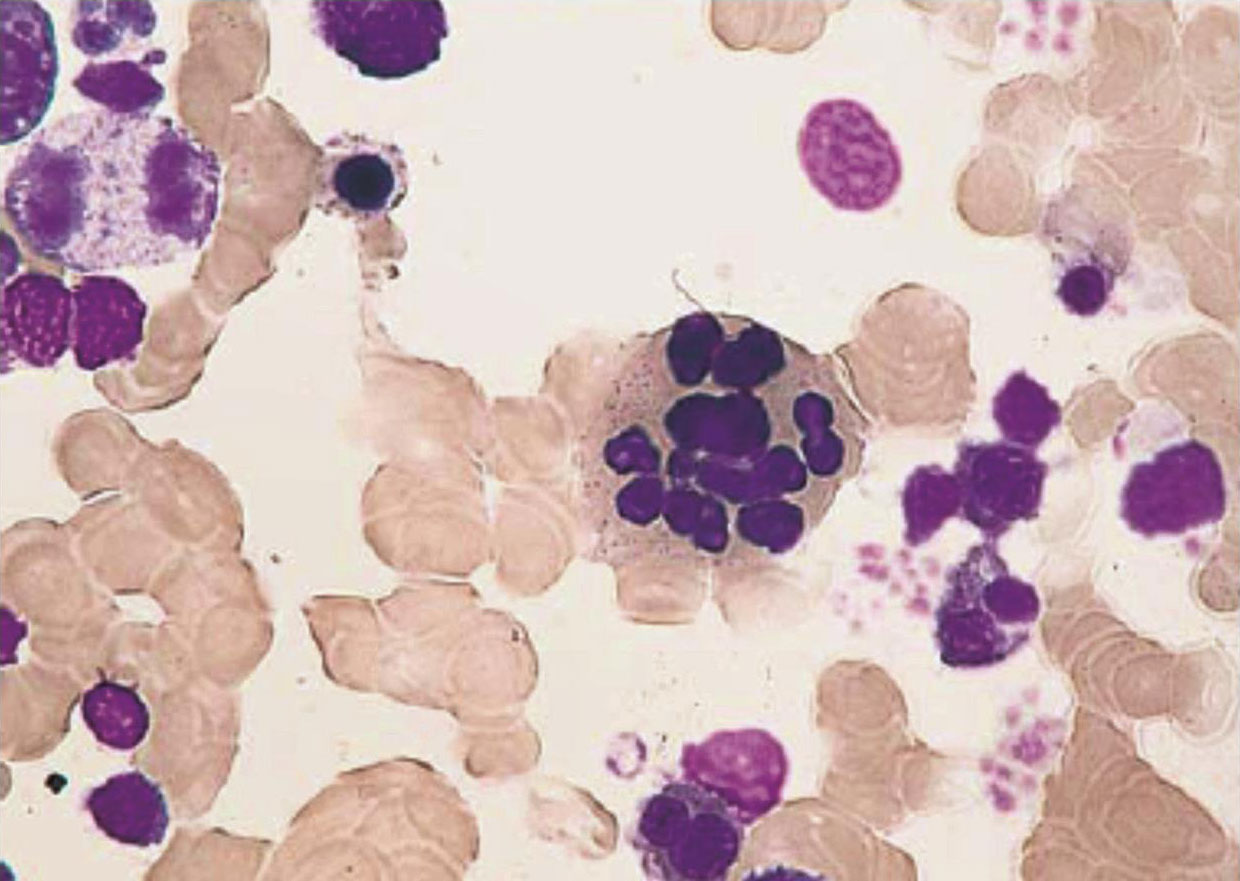
图16-2-17 先天性红细胞生成异常性贫血,骨髓涂片中多核的幼红细胞
CDA发病年龄差异很大,可在新生儿胎儿水肿时被诊断,也可在青少年发病。临床表现有间歇性黄疸、贫血、暗红色尿等。少数病例仅有高胆红素血症,而无贫血。慢性患者常伴胆石症。肝脾肿大程度不一。
根据细胞形态和血清学的改变分为Ⅰ、Ⅱ、Ⅲ三型,以CDA Ⅱ型最常见,约占病例的2/3,也有些病例难以归入三型中的任何一型,可称为变异型。
CDAⅠ型约占15%,为常染色体隐性遗传,基因定位于15q15.1-15.3。酸溶血试验阴性,诊断主要依据形态学改变。多以贫血、黄疸、肝脾大起病,可有指(趾)骨畸形等表现。贫血为大细胞性,骨髓红系增生伴巨幼样变。电镜下见幼红细胞核染色质呈海绵状,有染色质间桥。大多进展缓慢,少数需要输血,输血依赖者可考虑脾切除。EPO治疗一般无效。干扰素α在部分患者有效,特别是伴丙型肝炎的患者。如果有铁负荷过多的表现,应去铁治疗。
CDAⅡ型为常染色体隐性遗传,基因定位于20q11.2。本型除了骨髓红系形态学改变外,最大的特点是酸溶血试验阳性,患者红细胞在30%正常人血清中孵育,酸溶血试验呈阳性,但用自身血清做此试验为阴性,并且糖水试验亦为阴性,可与PNH鉴别。所以此型又称为:遗传性多核幼红细胞伴阳性酸溶血试验(hereditary erythroblastic multinuclearity with positive acidified test,HEMPAS)。另一血清学特点为红细胞表面存在胎儿i抗原,所以用抗i和抗I血清红细胞明显凝集。Ⅱ型患者红细胞膜蛋白有糖基化缺陷,SDS-PAGE电泳可见带3蛋白移动速率加快,而Ⅰ型和Ⅲ型不明显。患者贫血为正细胞性,有黄疸和肝脾大。大多进展缓慢,输血依赖者可脾切除。积极去铁预防继发性血色病。异基因骨髓移植可获治愈。
CDAⅢ型最少见,为常染色体显性遗传,基因定位于15q21-25。诊断主要依靠形态学,酸溶血试验阴性,但用抗i和抗I血清红细胞可以凝集。贫血为大细胞性,轻到中度。骨髓特征性表现为巨大的多核幼红细胞,直径可达50μm,最多可达12个核。与Ⅰ型不同,电镜下幼红细胞无海绵状染色质。以支持治疗为主。
主要参考文献
1.中国医师协会肾内科医师分会.肾性贫血诊断与治疗中国专家共识(2014修订版).中华肾脏病杂志,2014,30(9):712-715.
2.Goldman L,Schafer AI.Goldman's Cecil Medicine.25th ed.Philadelphia:Elsevier Saunders,2016,1064-1067.

