第三章
白血病和相关疾病
第一节
白血病概述

许小平
白血病(leukemia)是一组异质性恶性克隆性疾病,系早期造血前体细胞突变导致的造血系统恶性肿瘤。白血病的主要病理生理特征为异常血细胞(即白血病细胞)在骨髓及其他造血组织中大量增生,浸润各种组织,而正常造血功能受到抑制,正常血细胞生成减少,并产生相应的临床表现。
一、分类和分型
(一)按细胞分化程度分类
可将白血病分为:①急性白血病(acute leukemia):骨髓及外周血中以异常的原始及幼稚细胞为主,一般超过20%。病情发展多比较迅速。②慢性白血病(chronic leukemia):骨髓及外周血中以异常的较成熟细胞为主,其次为幼稚细胞,原始细胞常不超过10%~15%。大多表现为疾病进展缓慢。
(二)按白血病细胞的形态和细胞化学特征分类
1976年FAB(法-美-英)协作组将急性白血病分成急性淋巴细胞白血病(acute lymphoblastic leukemia,ALL)和急性髓系白血病(acute myeloid leukemia,AML)两类。FAB分类以形态学为主,自1976年发表以来,因方法简易实用,对白血病的治疗和和估计预后有一定价值。后又经多次修订完善,得到世界各国血液病学家的普遍认可,成为世界卫生组织(WHO)造血与淋巴组织肿瘤分类及诊断标准颁布以前国际上统一的分型方法,至今仍有重要影响。FAB分类将ALL按原始淋巴细胞的大小及形态学分L 1 、L 2 和L 3 三个亚型,急性髓系白血病分成M 0 至M 7 八个亚型,其中M 0 、M 1 、M 2 、M 3 系按髓系白血病细胞的分化程度依次分型,M 4 为急性粒单核细胞白血病(acute myelomonocytic leukemia,AMMoL),简称急粒单,M 5 为急性单核细胞白血病(acute monocytic leukemia,AMo L),简称急单,M 6 为急性红白血病(acute erythroid leukemia,AEL),M 7 为急性巨核细胞白血病(acute megakaryoblastic leukemia,MKL)。我国1986年在天津召开全国白血病分类、分型讨论会,以FAB分型方法为基础将 AML分成 M 1 、M 2a 、M 2b 、M 3a 、M 3b 、M 4a 、M 4b 、M 4c 、M 4 E 0 、M 5a 、M 5b 、M 6 、M 7 等亚型。
慢性白血病分为慢性淋巴细胞白血病(chronic lymphcytic leukemia,CLL)、慢性粒细胞白血病(chronic myelogenous leukemia,CML)、慢性粒-单核细胞白血病(chronic myelomonocytic leukemia,CMML)、慢性中性粒细胞白血病(chronic neutrophilic leukemia,CNL)、毛细胞白血病(hairy cell leukemia,HCL)和幼淋巴细胞白血病(prolymphocytic leukemia,PLL)等。
少见和特殊类型白血病包括低增生性白血病(hypoplastic leukemia)、髓系肉瘤(myeloid sarcoma)、嗜酸性粒细胞白血病(eosinophilic leukemia)、嗜碱性粒细胞白血病(basophilic leuke-mia)、肥大细胞(或组织嗜碱细胞)白血病(mast cell leukemia)、成人T细胞白血病(adult T cell leukemia)、浆细胞白血病(plasma cell leukemia)、毛细胞白血病、急性未明系列白血病(acute leukemia of ambiguous lineage)和急性全髓增殖症伴骨髓纤维化(acute panmyelosis with myelofibrosis)等。
(三)MICM分类
应用单克隆抗体进对急性白血病进行免疫学分型,可将ALL分成6个亚型;早B前体型、普通型、前B型、B细胞型、早T前体型和T细胞型。但髓系白血病细胞在不同成熟阶段的表面抗原并不呈一致性顺序出现,所以对不同亚型的髓系白血病进行免疫学分类尚有困难,目前主要将 MPO以及CD13、CD33、CD117等用于诊断髓系来源白血病的重要参考指标。由于白血病细胞可出现细胞系间反常抗原表达,已发现了有两系或两系以上系标记并存的恶性造血细胞;成人ALL中有23%表达髓系抗原,AML中有20%表达淋巴细胞相关分化抗原。
20世纪90年代起,人们认识到在白血病这组高度异质性的疾病中,细胞分子遗传学的标志性改变往往较形态学、细胞化学及免疫标志的变化更能深刻反映疾病本质,因此,国际上白血病分型逐步倾向于形态学(M)、免疫学(I)和遗传学(C)以及分子生物学(M)方法综合评估的MICM分类法。2001年正式公布的WHO造血和淋巴组织肿瘤分类和诊断标准(简称WHO分类标准)即是应用MICM分类法的典范。该分类标准于2008年第4版又作了重要修订。WHO分类中疾病实体之间界限较FAB分类更为清晰,解决了某些单凭形态难以分型或易混淆白血病的诊断,目前已被国内外血液和肿瘤工作者广泛采用。WHO分类标准将急性白血病分为AML、ALL和系列不明急性白血病三大类(表16-3-1~表16-3-3)。将AML和ALL中伴有重现性遗传学异常单独分开不仅有助于了解各型急性白血病的发病机制,而且对临床医生判断预后及选择不同治疗策略有重要参考价值。按WHO分类,原始淋巴细胞 白血病(lymphoblastic leukemia,ALL)和原始淋巴细胞淋巴瘤(lymphoblastic lymphoma,LBL)同属一种疾病实体,称 ALL/LBL。FAB分类中的ALL-L 3 已命名为Burkitt淋巴瘤/白血 病(Burkitt lymphoma/leukemia),归入成熟B细胞肿瘤,不应称B-ALL;将慢性白血病中慢性淋巴细胞白血病/小淋巴细胞淋巴瘤(chronic lymphocytic leukemia/small lymphocytic lymphoma,CLL/SLL)、B-幼淋巴细胞白血病(B-PLL)、毛细胞白血病(HCL)归入成熟B细胞肿瘤;将T-幼淋巴细胞白血病(T-PLL)、T-大颗粒淋巴细胞白血病(T-cell large granular lymphocytic leukemia,T-LGL)、侵袭型 NK细胞白血病(aggressive NK cell leukemia)、成人 T细胞白血病/淋巴瘤(adult T-cell leukemia/lymphoma,ATLL)归入成熟T细胞和NK细胞肿瘤;将慢性粒细胞白血病(CML)、慢性中性粒细胞白血病(CNL)、慢性嗜酸性粒细胞白血病(chronic eosinophilic leukemia,CEL)归入骨髓增殖性肿瘤(MPN);将慢性粒-单核细胞白血病(CMML)归入骨髓增生异常/骨髓增殖性肿瘤(myelodysplas-tic/myeloproliferative neoplasms,MDS/MPN)。
表16-3-1 WHO(2008)有关AML分类

表16-3-2 WHO(2008)有关ALL分类

表16-3-3 WHO(2008)有关系列不明急性白血病分类
二、流行病学
白血病是一种常见的恶性肿瘤,占癌症总发病数的3%~5%左右。西方国家统计,白血病总年发病率8~10/10万,近30年来都稳定在此水平,推算全世界每年有新病例20万~25万,在同一时期的估计病例数可达50万以上。
(一)发病率与死亡率
我国来自全国肿瘤登记中心收集到的32个省、自治区、直辖市261个肿瘤登记处提供的肿瘤登记资料,2012年全国白血病总发病率以及男、女性发病率均在10大常见肿瘤之外,但农村地区发病率居常见肿瘤的第10位,为5.11/10万,年龄标准化后的发病率4.50/10万。
2012年全国白血病总死亡人数5.2万;死亡率3.85/10万,年龄标准化后的死亡率3.13/10万,位于10大常见肿瘤的第9位。男性死亡率4.41/10万,年龄标准化后的死亡率3.70/10万,位于第7位;女性死亡率3.26/10万,年龄标准化后的死亡率2.58/10万,位于第7位。城镇地区死亡率4.05/10万,年龄标准化后的死亡率3.17/10万,位于第8位;农村地区地区死亡率3.63/10万,年龄标准化后的死亡率3.09/10万,位于第9位。
据美国统计,1970—1994年年龄标化白血病死亡率,白人男性8.80/10万,女性5.16/10万。最新根据SEER(Surveillance,Epidemiology,and End Results)资料的研究估计,美国2016年新增白血病患者60 140例,其中AML 19 950例,ALL 6590例,CML 8220例,CLL 18 960例,其他白血病6420例。死于白血病的患者数估计为24 400例,其中AML 10 430例,ALL 1430例,CML 4660例,CLL 1070例,其他白血病6810例。白血病死亡率占所有癌症死亡率的4%。
CML在美国大约占所有白血病的1/5,每年新诊断的患者1/10万~2/10万,全国总数为4800~5000例。近10年发病率并无明显变化。但由于治疗水平提高,年死亡率从2000年以前的10%~20%下降到目前的1%~2%。因此,预测到2030年,美国的CML患者数将从2000年以前的15 000~20 000例增加到180 000例。
CLL是西方国家最常见的白血病类型。占所有白血病的1/3左右。是CML的2倍。CLL诊断后5年存活率为80.8%,因此,美国估计现有105 634例患者存活。
白血病发病率世界范围的地理学差异研究尚无得出可信服的结论。根据1997年五大洲癌症发病率统计,白血病发病率最高的是北美、澳大利亚和新西兰的白人;其次是北欧、西欧;南欧和以色列犹太人为中等水平;中国、日本、印度为低水平,美国非西班牙裔白人男性年龄校正发病率高达14.1/10万人年,女性为8.3/10万人年。我国白血病的发病率与亚洲国家相近而明显低于欧美国家。
白血病发病率的种族差别只有CLL较肯定,而其他类型白血病并不明显。CLL在亚洲国家如中国和日本的发病率仅为美国和其他西方国家的10%。
国外多数统计资料认为从全球范围看,自60年代以来,白血病的发病率基本保持平稳。国内白血病的发病率是否逐年升高未有肯定的结论。
(二)性别和年龄发病率
据各地区、各年代白血病的性别发病率调查,男女之比为1∶1~1.6∶1。我国1986—1988年调查资料表明男性白血病发病率为2.98/10万,女性为2.52/10万,男性发病率略高于女性,尤其是青少年和老年人发病率的性别差别更明显。CML和CLL各年龄组都是男性发病率高于女性。
观察白血病年龄的发病率曲线,发现在5岁以下及15~20岁间有两个小高峰,在40岁以后随年龄增加发病率逐渐升高,高峰年龄在60岁以后。各型白血病发病年龄不尽相同,AML初诊中位年龄67~71岁,20岁以下为6.3%,发病率随着年龄增加而增高,从青少年的1.3/10万增加至70~80岁年龄段的15/10万。ALL发病率存在成人(>19岁)、儿童(2~19岁)以及婴儿(1岁以内)三个明显的年龄段差异,其中以2~10岁之间发病率最高。ALL是15岁以下儿童中最常见的恶性肿瘤,大约占儿童癌症死亡者的1/3。年大约有新诊断的儿童ALL患者2400例、成人ALL患者5500例、婴儿ALL患者200例。年龄调整后发病率为1.34/10万。CML发病率每年在1.3~1.6/10万之间,占新诊断成人白血病的15%~20%,但5~20岁年龄段的患者仅占所有CML的10%,中年后发病率增高,美国2004—2009年CML诊断的中位年龄为64岁。CLL发病率在中年后随着年龄增加呈上升趋势,在80岁人群的年发病率高达20/10万。根据SEER 2000—2005年的数据分析,美国CLL中位诊断年龄为72岁,35岁以下患者<1%,45岁以后发病率逐步升高,75~84岁达最高峰,年龄调整后的发病率为4.2/10万。
(三)各型白血病的发病率与构成比
据美国统计各型白血病的构成比为:AML 34%,CLL 28%,CML 13%,ALL 11%,其余为其他类型白血病。日本由于CLL发病率低(构成比2.5%),因此急性白血病占70%以上。在儿童中绝大多数为急性,慢性仅占2.7%~5.7%,几乎所有儿童的慢性白血病均为慢粒。根据我国1986—1988年调查资料,各型白血病的发病率以AML最高(1.62/10万),ALL次之(0.69/10万),CML(0.36/10万)、CLL(0.05/10万)和特殊类型(0.03/10万)最低。1999年上海市白血病协作组统计2867例急性白血病FAB分型资料,各亚型构成比:AML61.6%,ALL35.4%,其他3%;其中儿童分别为31.3%、66.3%、2.5%;青壮年分别为 70.6%、26.8%、2.6%;老年分别为77.5%、17.0%、5.5%。ALL中L1占43.6%,L2占48.4%,L3占8%。AML中 M 1 7%、M 2 24.9%、M 3 22.7%、M 4 15.1%、M 5 26.5%、M 6 3.1%、M 7 0.7%。
三、病因和发病机制
人类白血病的确切病因至今未明。许多因素被认为与白血病的发病有关。病毒可能是主要的因素,此外尚有遗传因素、放射、化学毒物或药物等因素。某些染色体的异常与白血病的发生有直接关系,染色体的断裂和易位可使癌基因的位置发生移动和被激活,染色体内基因结构的改变可直接引起细胞发生突变,免疫功能的降低则有利于白血病的发生。
(一)病毒
早已证实,C型RNA肿瘤病毒或称逆转录病毒是哺乳类动物(如小鼠、猫、牛、绵羊)和灵长动物自发性白血病的病因,这种病毒能通过内生的逆转录酶按照RNA顺序合成DNA的复制品,即前病毒,当插入宿主的染色体DNA中后可诱发恶变。肿瘤病毒携有病毒的原癌基因(v-onc),大多数脊椎动物(包括人)的细胞基因体内也有与v-onc同源的原癌基因。v-onc被整合入宿主细胞的基因后可使邻近的基因发生恶变。逆转录病毒的感染也可致原癌基因激活,成为恶性转变的基因,导致靶细胞恶变。进入体内的病毒基因即使不含有v-onc,如果改变了基因的正常功能,也有可能引起白血病。
人类白血病的病毒病因研究已有数十年历史,但至今只有两种少见类型的白血病是和病毒关系较为肯定:成人T细胞白血病肯定是由逆转录病毒引起;EB病毒属DNA病毒,被认为和Burkitt白血病(成熟B-ALL)发病有关。1976年日本人发现了成人T细胞白血病/淋巴瘤(ATL),以后流行病学调查,发现在日本西南部、加勒比海区域及中部非洲为高发流行区。1980年在ATL细胞系中发现ATL相关抗原,并在电镜下发现了病毒颗粒。美国的Gallo和日本的日昭赖夫分别从患者培养细胞株中分离出C型逆转录RNA病毒,命名为人类T细胞白血病/淋巴瘤病毒(HTLV-Ⅰ)和成人T细胞白血病病毒(ATLV),以后证实HTLV-Ⅰ和ATLV是一致的。这是对人类白血病病毒病因研究的重大贡献。在ATL细胞中发现有HTLV-Ⅰ前病毒DNA,ATL患者血清中可检出HTLV-Ⅰ抗体。ATL的高发区也是HTLV-Ⅰ感染的高发区。血清流行病学调查表明,日本ATL流行区40岁以上的健康人群中HTLV-Ⅰ抗体阳性率达6%~37%。而非流行区抗体阳性率仅0~0.015%。HTLV-Ⅰ具有传染性,可通过乳汁母婴传播,通过性交和输血传播,除ATL外其他类型白血病尚无法证实具有传染性。我国东南沿海的地理位置和气候条件与日本西南部相似,并且日本与我国相邻,两国交往密切。为了弄清楚这些地区是否也有本病流行,中国预防医学科学院病毒学研究所等单位曾调查我国28个省、市、自治区人群的10 013份血清标本,发现8例HTLV-Ⅰ抗体阳性,其中3例日本人,2例为我国台湾人,2例中国妇女系上述阳性者的妻子,1例系我国海员,常在日本港口居住。说明病毒可由与日本人密切接触而传播。1989年吕联煌等在福建沿海地区发现HTLV-Ⅰ小流行区。共调查518人,包括白血病患者210人,发现2例成人T淋巴细胞白血病患者和5例血清HTLV-Ⅰ抗体阳性者,其中1例ATL,患者的妻子和次子血清HTLV-Ⅰ抗体均为阳性,1例毛细胞白血病患者血清HTLV-Ⅰ抗体也呈阳性。1982年Kalyanaraman从1例T细胞型毛细胞白血病患者的细胞株中分离出HTLV,他认为该例患者的病毒有别于以前分离出的HTLV,称为HTLV-Ⅱ型病毒,提出毛细胞白血病的病毒是HTLV-Ⅱ。
(二)遗传因素
遗传因素和某些白血病发病有关。白血病患者中有白血病家族史者占8.1%,而对照组仅0.5%。近亲结婚人群ALL发病率比期望值高30倍。单卵双胎者如一人患白血病,另一人患白血病的机会为20%,并且双胎可得同型白血病。家族性白血病占白血病病例总数的7%,国外已报道100多例。国内张氏和程氏曾报道11户22例和8户16例家族性白血病,主要为父母与子女或兄妹之间。1985年以来我国报道先天性白血病(congenital leukemia)11例,其中有急粒4例,急粒单1例,急单3例,急淋3例。在白血病的第1代家族中患白血病者,急性白血病占2%,CLL占3.5%,CML只占0.8%,故CLL的家族性较显著。
某些染色体有畸变、断裂或DNA修复有缺陷的遗传性疾患常伴较高的白血病发病率,如唐氏综合征(Down syndrome)、先天性血管扩张红斑症即布卢姆综合征(Bloom syndrome)、共济失调毛细血管扩张症(ataxia telangiectasia)、遗传性8号染色体三体综合征和范科尼贫血(Fanconi anemia)等。Down综合征有21号染色体三体异常,其发生急性白血病的危险性比一般人群高20倍,且发病比其他儿童为早(中数发病年龄2~3岁 vs .5~6岁),并且常伴AML-M 7 。Bloom综合征易有染色体断裂,其发生急性白血病的危险性可高达50%。Fanconi贫血是常染色体隐性遗传性疾病,具有自发性染色体断裂,患者及其家族中白血病的发病率甚高。上述遗传性疾患不仅有染色体的异常,并且其体细胞在体外病毒作用下有很高的恶变率。先天性无丙种球蛋白血症虽未检出有染色体异常,但均有细胞免疫及体液免疫的缺陷,严重联合免疫缺陷者白血病的发生率也很高。
白血病和HLA抗原型别有某种联系尚有争议。有报道ALL常伴HLA-A 2 和A 9 ,提示遗传因素与白血病的发病有某种联系,但对大多数白血病而言,白血病并不是遗传性疾病。
(三)放射因素
电离辐射有致白血病作用,其作用与放射剂量大小、放射部位及年龄有关。短期内较大剂量照射、全身和放射野较大的照射,特别是骨髓受到照射,可导致骨髓抑制和免疫抑制,照射后数月可观察到染色体的断裂和重组。尤其是年幼患者危险性较高。放射可诱发AML、ALL和CML,但未见CLL,并且发病前常有一段骨髓抑制期,其潜伏期为2~16年。1945年日本广岛和长崎遭原子弹袭击后幸存者中发生白血病数比未经辐射者高数十倍。在广岛和长崎严重辐射地区的白血病发病率分别较未辐射地区高30倍和17倍,到1978年止发现了135例急性白血病和53例CML,并发现其发病数和受照剂量有关。放疗也可致白血病,强直性脊柱炎患者放疗后白血病发生率较一般人群高十几倍,并且与剂量有关:累积剂量>20Gy,其相对危险度为14.4,<5Gy的相对危险度为4.2。真性红细胞增多症采用随机对照设计,不同治疗方法观察以后发生白血病危险性:放血治疗1%, 32 P治疗6%,苯丁酸氮芥治疗11%。说明化疗致白血病作用高于放疗。职业性长期照射也可致白血病,据我国1950—1980年调查,临床X线工作者白血病发病率为9.61/10万(标化率9.67/10万),而其他医务人员为2.74/10万(标化率2.77/10万)。对1950—1990年间放射诊断工作者白血病的危险性进行分析,发现其白血病发病率为对照组的2.25倍。诊断性照射是否会致白血病尚无确切的根据,但妊娠妇女胎内照射可增加出生后的小儿白血病的危险性。超低频非离子化电磁场也可致白血病,但作用甚小。
(四)化学因素
苯的致白血病作用比较肯定。苯的毒性作用和累积剂量有关,1~10ppm可致染色体损伤,124~200ppm有致白血病危险。我国工厂接触苯工人调查,其发生白血病危险性是一般人群4~7倍,平均潜伏期11.4年,暴露在≥25ppm,危险性最高,并有剂量反应关系。据报道40例苯致白血病的类型包括急粒(15例)、红白血病(7例)、白血病前期(7例)、急淋(4例)、急单和急粒单(3例)、CML(2例)、早幼粒及不能分类白血病各1例,未见CLL。苯致急性白血病以AML和红白血病为主,红白血病占相当比例,值得引起注意;并且在出现白血病临床表现常有一阶段骨髓抑制期。日常生活中接触含苯物质如染发剂、家庭装修材料是否与白血病发病有关,尚存在争议。
烷化剂、拓扑异构酶Ⅱ抑制剂(依托泊苷、替尼泊苷、多柔比星、柔红霉素、米托蒽醌等)可致继发性白血病也较肯定。烷化剂可致点突变,激活癌基因致染色体异常,拓扑异构酶Ⅱ抑制剂可致DNA复制关键酶缺失,导致染色体异常。Curtis调查了美国1973—1980年诊断为癌症的44万例患者,其中单用化疗者发现了47例继发性白血病,显著高于预期数,其中AML 34例。多数继发性白血病是在原有淋巴系统恶性肿瘤和易产生免疫缺陷的恶性肿瘤经长期烷化剂治疗后发生,乳腺癌、卵巢癌和肺癌化疗后也易发生继发性白血病。文献中搜集到22 986例霍奇金淋巴瘤,经化疗后0.17%~2.4%发生白血病,发病间隔2~8年。2861例多发性骨髓瘤,经化疗后0.6%~7.9%发生白血病,发病间隔2.5~6年。烷化剂应用后发生继发性白血病,潜伏期常为5~9年,老年人危险性增加,潜伏期可缩短,发生白血病前可有MDS前期,常呈骨髓低增生和纤维化,白血病类型多数是AML,很少有Auer小体,累及染色体异常多为复杂核型、单体7(-7)、del(5q)和-5。拓扑异构酶Ⅱ抑制剂致继发白血病潜伏期短(6个月~5年),无MDS前期,多数为AML-M 4 和 M 5 ,累及染色体异常为11q23和21q22。基于以上研究,WHO(2008)造血与淋巴组织肿瘤分类标准将治疗相关髓系肿瘤作为一种独立的疾病实体列出。
国内曾陆续报道过乙双吗啉所致继发性白血病数百例。该药是乙亚胺的衍生物,用于治疗银屑病,是一种极强的致染色体畸变的物质。服乙双吗啉后1~7年发生白血病(中位数4年),白血病类型主要为AML,其中以 M 3 最多,可能乙双吗啉作用于15号和17号染色体。与白血病发病可能有关的药物还有氯霉素和保泰松,但尚无非常肯定的结论。
(五)其他
曾有研究者怀疑吸烟、饮酒及长期过度肥胖亦与白血病发病有关,但至今尚无说服力较强的证据。
主要参考文献
1.林果为,欧阳仁荣,陈珊珊,等.现代临床血液病学.上海:复旦大学出版社.2013,807-837.
2.Swerdlow SH,Campo E,Harris NL,et al.WHO Classification of Tumors of Haematopoietic and Lym phoid Tissues.4th ed.Lyon:IARC,2008,110-178.
3.Lee GR,Foerster J,Luken J,et al.Wintrobe's Clinical He-matology.13th ed.Baltimore:Williams&Wilkins,2014,1548-1928.
4.Hoffman R,Benz Ir EJ,Siberstein LE,et al.Hematology:Basic principles and Practice.6th ed.Philadephia:Elsevier saunders,2013,853-881.
5.Goldman L,Schafer AI.Goldman-Cecil Medincine.25th ed.Philadelphia:Elsevier Saunders,2016,1239-1257.
6.Siegel RL,Miller KD,Jemal A.Cancer statistics,2016.CA Cancer J Clin,2016,66(1):7-30.
第二节
急性白血病
许小平
一、临床表现
各类急性白血病的共同临床表现大多与正常造血细胞生成受抑和白血病细胞增殖浸润有关。正常造血细胞生成受抑可引起感染、发热、出血和贫血;白血病细胞增殖浸润可导致肝、脾、淋巴结肿大及其他器官病变。症状的缓急主要取决于白血病细胞在体内的增长速率和积蓄程度。
(一)感染
约半数以上患者以发热起病,当体温>38.5℃时常由感染引起。感染是急性白血病最常见的死亡原因之一。据上海市白血病协作组统计,初诊时46.1%的AML和42%的ALL患者有感染发热。国外494例急性白血病1894次发热分析显示,明确为感染者占64%,不明原因者35%,非感染性者仅占1%。急性白血病发生感染的机制:①中性粒细胞数量减少和功能缺陷:白血病细胞能抑制骨髓正常粒系祖细胞的生成,加上化疗药物对骨髓的抑制毒性,在诱导缓解期常发生显著的粒细胞缺乏症,极易并发各种细菌或真菌感染。中性粒细胞<1×10 9 /L时,感染机会中度增加;<0.5×10 9 /L时,显著易感染;<0.1×10 9 /L时,几乎都有严重的感染。感染的发生还和粒细胞缺乏持续的时间有关,超过2周者几乎都有严重感染,且真菌和原虫感染的危险性显著增高。粒细胞的趋化、游走、吞噬及杀菌功能降低,不能产生正常的炎症反应,感染不易局限。②免疫缺陷:化疗及应用糖皮质激素等可加重免疫紊乱。免疫球蛋白合成减少,补体缺乏,使机体对具有荚膜的细菌如肺炎链球菌或流感杆菌的防御能力显著减弱,加上细胞免疫功能减低,患者易发生范围广泛的各种病原体感染。③皮肤黏膜屏障破坏更有利于病原体的入侵。④院内感染:长期住院患者的感染半数系院内获得,细菌常呈耐药性。感染以咽峡炎、口腔炎最多见,肺部感染、肛周炎及肛周脓肿也常见。皮肤黏膜感染很少化脓,但易形成蜂窝织炎。胃肠道感染常是脓毒血症的主要来源。泌尿系感染时尿路刺激症状不明显;当白细胞<0.1×10 9 /L时,仅11%有脓尿。在发病早期,感染常由革兰阳性球菌如粪链球菌、金葡菌或表葡菌所引起;但长期反复抗生素治疗后体内菌群发生变化,加以肠道黏膜溃疡和肠壁白血病细胞浸润,此时革兰阴性杆菌感染较多见。细菌多数来自患者本身的肠道,其中50%以上系住院后获得,以硝酸盐阴性、肺炎和铜绿假单胞菌为多见,占感染死亡的75%,尤其是假单胞菌感染患者常出现典型的坏死性皮损,死亡率高。结核复发也有报道。真菌常为终末期感染,但也有发生在病程早期,尸检发生率占13%,以念珠菌及曲霉菌多见。急性白血病发生病毒感染时病情常较凶险,如麻疹或水痘易并发肺炎、脑炎等。病毒感染中巨细胞病毒(CMV)常见于急性白血病缓解期,尤其是儿童ALL。
(二)出血
40%~70%的患者起病时伴出血倾向。在未并发弥散性血管内凝血(DIC)者,出血的发生率为67%~75%,死于出血占10%~15%。并发的DIC患者几乎全部有出血,其中死于DIC者占20%~25%。AML有出血倾向(58%)者明显多于ALL(42%)。
出血的机制如下:①血小板减少:约95%的AL病例有血小板减少,是引起出血最重要的原因。皮肤淤点、淤斑和齿龈渗血最常见,可有鼻出血和月经过多。视网膜出血时可引起失明,蛛网膜下腔出血常引起突然死亡。血小板功能障碍并非是AL出血的主要原因。当血小板在20×10 9 /L以上时可无严重出血,但低于5×10 9 /L者常引起致命的出血倾向。如血小板在20×10 9 /L以上有严重出血常提示有其他机制参与出血,但某些AL可有血小板的黏附、聚集和释放功能异常,血小板膜糖蛋白Ⅰb和Ⅱb/Ⅲa异常,电镜观察常见α颗粒减少和体积变小。②血管壁损伤:由于白血病细胞浸润、感染内毒素以及大量化疗所引起。当白血病细胞数异常增多时,可使小动脉和小静脉内白血病细胞堆积,称白细胞淤滞,可发生出血。③凝血障碍:单个凝血因子缺乏较少见。凝血障碍常呈大块淤斑和血疱,伴有疼痛。内脏出血多见,如消化道、泌尿道、颅内出血。最常见的类型是DIC,AL并发DIC的发生率为7%~30%。急性早幼粒细胞白血病(APL)的出血机制较为复杂,以前多认为是白血病细胞颗粒中含有的促凝物质释放导致DIC的发生,现认为APL患者出血以原发性纤维蛋白溶解亢进为主。④抗凝物质增多:AL患者肝素或肝素类物质增多,发生率约占10%~15%。细菌感染释放有抗凝作用的多糖体,故感染可使出血加重。
(三)贫血
约2/3的AL患者在确诊时有中度贫血。贫血发生的机制为:①白血病细胞克隆能抑制正常多能造血干细胞以及红系祖细胞,并使红系祖细胞对红细胞生成素的反应性降低。白血病细胞破坏诱导红系生成的微环境等,从而使红系生成减少。②无效性红细胞生成:测定血浆和红细胞内放射性铁的转换以研究骨髓红系造血,发现白血病患者红系铁转换率正常或升高,但成熟红细胞的铁摄取量却显著降低,提示无效性红细胞生成,另外,某些类型的白血病患者伴幼红细胞增生异常,表现为巨幼样变和细胞分裂受阻。③溶血:明显溶血绝大多数见于淋巴细胞白血病。隐性溶血表现为对输血的要求明显增加,发生机制可能和免疫有关,少数可能有红细胞内在缺陷。DIC可伴微血管病性溶血性贫血。④其他:急慢性失血以及某些抗代谢化疗药物例如甲氨蝶呤(MTX)和阿糖胞苷(Ara-C)等可引起DNA合成障碍,导致巨幼细胞贫血。
(四)淋巴结和肝脾大
初诊时62.2%ALL患者、41%AML患者有淋巴结肿大,常见为浅表淋巴结肿大。淋巴结肿大以ALL为著。60%~80%的T-ALL有纵隔淋巴结肿大,严重者可引起气管、颈静脉压迫等症状。在AML中以M 4 及 M 5 发生淋巴结肿大多见,肝、脾大ALL较AML更为多见,据上海地区统计,初诊时ALL有60%的病例有肝大,47.9%有脾大,而AML仅31.8%的病例有肝大,20.9%有脾大。
(五)神经系统
中枢神经系统白血病(CNSL)以蛛网膜及硬脑膜浸润最高,分别为82%及78.6%,其次为脑实质(62%)、脉络丛(42%)及脑神经(22%),可发生在白血病活动期或缓解期。约有2%的急性白血病患者初诊时有中枢神经系统累及,如未进行中枢神经系统白血病的预防处理,则70%的ALL、20%~40%的儿童及5%的成人AML可发生中枢神经系统白血病。轻者可无症状或仅有轻微头痛,脑脊液压力增高。严重的才呈典型脑膜炎表现,但一般不发热。脑脊液检查可见压力增高,细胞数增多甚至发生混浊,蛋白增多,糖降低。利用细胞离心沉淀涂片染色检查,可检出白血病细胞。当周围血原始细胞显著增多(>50~75×10 9 /L)时,常可引起白细胞淤滞,多见于AML和CML的急变期。临床表现类同脑血管意外,患者有头痛、轻瘫,迅速进入昏迷,常致死亡。
(六)口腔及皮肤
白血病细胞浸润口腔黏膜可引起齿龈肿胀或巨舌等,多见于AML-M 5 及M 4 。白血病性齿龈炎常继发感染、出血,甚至发生继发性口干燥症。偶见急性白血病可首发于皮肤。皮肤浸润表现有白血病疹(leukemids)、结节、斑块和溃疡等。白血病疹呈淡紫色小丘疹,常有痒感,以AML-M 4 及M 5 为明显。活检或皮损印片有助于诊断。皮肤感染很多见,表现为蜂窝织炎,常呈大片状,迅速发展,最常见于面部,多由革兰阳性细菌所引起。病毒性皮炎常发生在化疗中或化疗后,以单纯疱疹及带状疱疹为多见。
(七)心脏和呼吸系统
急性白血病的肺部表现可由感染、浸润及白细胞淤滞等引起。初诊时有肺浸润者占5%,尸检中发现者占50%。肺浸润以AML常见,浸润多位于肺泡间隔,尤位于血管和小支气管周围,但引起肺动脉栓塞导致肺梗死者罕见。肺门和纵隔淋巴结肿大的发生率分别为27%和36%。因浸润出现渗出性胸膜炎及血性胸腔积液者多见于ALL,亦可见于AML-M 5 。肺部浸润的X线表现可呈弥漫性网状结节样改变,也可散在分布,和感染并存可呈片状阴影。肺部血管的白细胞淤滞可导致呼吸窘迫综合征,主要见于高白细胞急性白血病患者,病死率高。心肌及心包浸润的尸检报告可达35%,多见于ALL,有临床症状者仅5%,可表现为心肌炎、心律失常、心衰,偶有心包炎表现。
(八)骨和关节
骨痛及胸骨下端压痛常见。初诊时有骨、关节症状者ALL占11%,AML占2%。慢粒急变常有显著骨痛。骨痛可由于:①白血病细胞影响骨膜;②不明原因的骨梗死和骨髓坏死;③高尿酸血症致痛风发作;④溶骨性髓细胞肉瘤等。骨骼病变可通过X线摄片、骨扫描及骨MRI等检查确诊。
(九)性腺
性腺浸润占4%~27%,约2%的ALL初诊时即有睾丸白血病。由于对中枢神经系统白血病的有效防治,使睾丸白血病成为第二个髓外复发的部位。尤以白细胞明显增高者以及ALL更易发生。睾丸白血病可无症状,常呈双侧或单侧弥漫性肿大,质硬,不透光,可经局部穿刺或活检证实。卵巢白血病症少见。阴茎异常勃起偶见于急性白血病患者,可能和海绵体内白血病细胞栓塞有关。
(十)其他
约25%的患者在确诊为白血病时胃肠道已有白血病浸润,但临床表现少见;即使有症状也与浸润程度不相称,表现为腹痛、腹泻、胃肠道出血、黏膜炎症、肠梗阻等。白血病肾脏浸润率可达52%。白血病细胞可浸润甲状腺、胰腺、下丘脑和神经垂体,且可并发糖尿、低血糖或尿崩症等。低血糖系外周血大量白血病细胞“窃取”血糖所致。急性白血病患者的生化代谢紊乱常是多因素的,化疗可使之加重,造成症状的复杂化,严重者可致死,故需及时纠正。高尿酸血症是AL最常见的代谢紊乱。由于白血病细胞的高代谢状态,故尿酸可增高,尤其当诱导缓解化疗后白血病细胞大量崩解,使血浆尿酸浓度显著增高。大量尿酸由尿中排泄可导致严重肾病,甚至急性肾衰竭。急性白血病患者的电解质紊乱变化多端,无一定规律性。低钠血症较常见,可由于原发性或化疗药物如环磷酰胺、长春新碱所致的继发性抗利尿激素分泌过多综合征而引起。高钾血症在白血病细胞大量崩解时常见,甚至可致心搏骤停;低钾血症可见于AML-M 4 及M 5 ,因这类白血病患者的血清溶菌酶增高导致肾小管损害。抗生素引起的肾病和肠道功能紊乱也可引起低钾。高钙血症的出现常提示预后不佳,患者出现乏力、嗜睡、恶心、烦渴等精神症状,常伴骨痛、骨质疏松、溶骨性病变和病理性骨折。高钙血症的多尿及排钾增多可引起代谢性碱中毒,低钙血症也是白血病化疗中的严重并发症。高镁血症常见于白血病活动期。代谢性酸中毒常由于乳酸积聚所引起,多见于急性白血病活动期,因大量白血病细胞的无氧糖酵解所致;由并发深部真菌感染等引起者亦有报道。急性白血病化疗后大量白血病细胞杀伤,细胞内容物大量释放入血可引起急性溶瘤综合征,出现高磷、高钾、低钙、高尿酸血症、少尿、急性肾衰竭等,可致患者迅速死亡。
二、实验室检查
(一)血常规
急性白血病初诊时,多数病例外周血有不同程度的血红蛋白及红细胞减少,据统计血红蛋白测定的范围自17~147g/L。贫血大多数呈正常细胞性,仅少数有成熟红细胞大小不等、嗜碱性点彩、多染性红细胞及出现幼红细胞,半数病例网织红细胞计数偏低。白血病可引起红细胞血型抗原的减弱,造成血型鉴定的困难。急性白血病初诊时外周血白细胞计数可降低、正常、增高或显著增高。约50%的AML和30%的ALL患者白细胞计数可<5×10 9 /L,甚至可<1×10 9 /L;也有>100×10 9 /L,称为高白细胞急性白血病,占所有急性白血病的8.5%。约有5%的AML,9%儿童ALL和17%成人ALL发生高白细胞急性白血病,尤多见于T细胞ALL和AML-M 5 。高白细胞急性白血病病情凶险,早期病死率高,缓解率低,预后差。外周血白细胞分类,最主要的发现是被累及的血细胞系列的原始和幼稚细胞百分比显著增多,范围可从5%~100%,但白细胞不增多性白血病患者,外周血中可仅有极少量甚至没有原始细胞或幼稚细胞出现。急性白血病患者初诊时均有不同程度血小板减少,据统计52.4%患者<60×10 9 /L。
(二)骨髓象
急性白血病初诊时骨髓象绝大多数呈增生活跃、明显活跃或极度活跃,分类中最主要的特征是被累及的血细胞系列有原始和幼稚细胞大量增生,而正常造血细胞如幼红细胞和巨核细胞则明显受抑制。据统计,增生极度活跃者占45.4%,明显活跃占30.2%,活跃占20.6%,增生减低占3.8%;后者多见于AML。约有10%的AML骨髓活检中显示增生降低,称为低增生性急性白血病。据统计,分类中原始细胞平均占64.4%,最低占10%,最高占99.2%。
白血病细胞具有共同的形态特点:大小不一,多数体积增大,核质比值增大,细胞核形态不规则,常有异形,核染质粗糙,分布不均,核仁较正常原始细胞为大且显著;核分裂象多见,核质发育失调,胞核发育常落后于胞质,细胞分化停滞在原始细胞或幼稚细胞(早幼)阶段,而趋向于成熟的细胞极少见,呈所谓“裂孔”现象。Auer小体可见于部分AML,一般不出现在ALL中。CML急变期找到Auer小体纯属罕见。
(三)细胞化学染色
细胞化学染色在急性白血病的分型诊断中有重要意义。①ALL的细胞化学染色特征:过氧化酶(POX)、苏丹黑B(SB)和氯化醋酸AS-D萘酚酯酶(AS-D-CE)均呈阴性反应;醋酸AS-D萘酚酯酶(AS-DAE)阴性或弱阳性;α-醋酸萘酚酯酶(α-NAE)大多阴性,一些细胞可呈局灶性阳性,少数病例有局灶性强阳性反应;PAS染色在部分病例的部分细胞中呈块状或颗粒状阳性,而无弥漫性着色;酸性非特异性酯酶(ANAE)和酸性磷酸酶(ACP)呈阴性或弱阳性反应。T细胞ALL的ANAE、ACP及末端脱氧核苷酸转移酶(TdT)的活性都显著增高;B细胞ALL的ACP、ANAE及TdT均为阴性反应。FAB协作组规定ALL可有3%原始细胞POX染色可呈阳性,因此POX阳性原始细胞>3%可作为ALL和急粒的鉴别点。其实ALL的3%POX阳性原始细胞并非是白血病原始细胞,而是正常的原粒和早幼粒细胞。②急粒细胞化学染色的特征:POX和SB染色对分化差的原粒细胞呈阴性反应,分化好的呈阳性反应,其强弱程度各异,M 1 型以阴性或弱阳性反应多,M 2a 和M 3 型以强阳性为多,Auer小体也呈阳性;AS-D-CE染色呈特异性阳性反应;非特异性酯酶(NSE)可呈阳性反应,但不被NaF抑制或抑制率<50%;中性粒细胞碱性磷酸酶(NAP)明显减少或消失。PAS染色根据白血病细胞的分化程度可呈阴性反应或呈弥漫性淡红色反应,M 3 型呈弥漫性红色反应。③急单细胞化学染色的特征:POX和SB染色时原幼单核细胞呈阴性或弱阳性反应;NSE呈阳性或强阳性反应,可被NaF抑制,抑制率>50%;AS-D-CE呈阴性反应,偶见弱阳性反应;NAP积分增高;血、尿溶菌酶活性显著增高。④急粒单细胞化学染色的特征:具有上述两系细胞的特征,并且过氧化酶-溶菌酶(POX-Lz)双重染色时Lz活性>POX,AS-D-CE和AS-D-E双重染色时两类不同细胞可显示两种不同的染色。⑤红白血病的幼红细胞PAS染色呈阳性反应,且多为颗粒或块状分布。
(四)免疫表型检查
按照T细胞分化模式,在淋巴系干细胞阶段仅有CD34、HLA-DR及TdT表达,继而出现CD7,同时胞质中开始表达CD3,标志着发育至幼稚胸腺细胞阶段,此时部分细胞可出现CD5、CD2;到皮质胸腺细胞期,CD1、CD4、CD8共同表达;髓质胸腺细胞和外周血T细胞一样,CD1消失,CD4或CD8在不同细胞上独立表达,胞膜上出现T细胞抗原受体复合物CD3标志。按照B细胞分化过程,其抗原表达继淋巴系干细胞之后,B系祖细胞便出现CD19,胞质中CD10开始表达;早前B细胞期CD34、TdT消失,膜CD10及胞质CD22出现;进入前B细胞期,Cyμ链、CD22、CD20均已表达;SmIg为成熟B细胞标志。按照髓系(粒-单系)细胞的分化过程,CD33和CD13是髓系发育成熟全过程均存在的抗原;CD34在髓系祖细胞表面出现,分化至原粒细胞逐渐消失;HLA-DR存在于CFU-GM和各期单核细胞上;到幼稚及成熟期,粒、单核细胞表面出现CD11b,粒系同时有CD15,单核细胞则表达CD14。细胞表面免疫学标记对白血病分型诊断意义见表16-3-4。
表16-3-4 细胞表面免疫学标记对白血病分型诊断意义
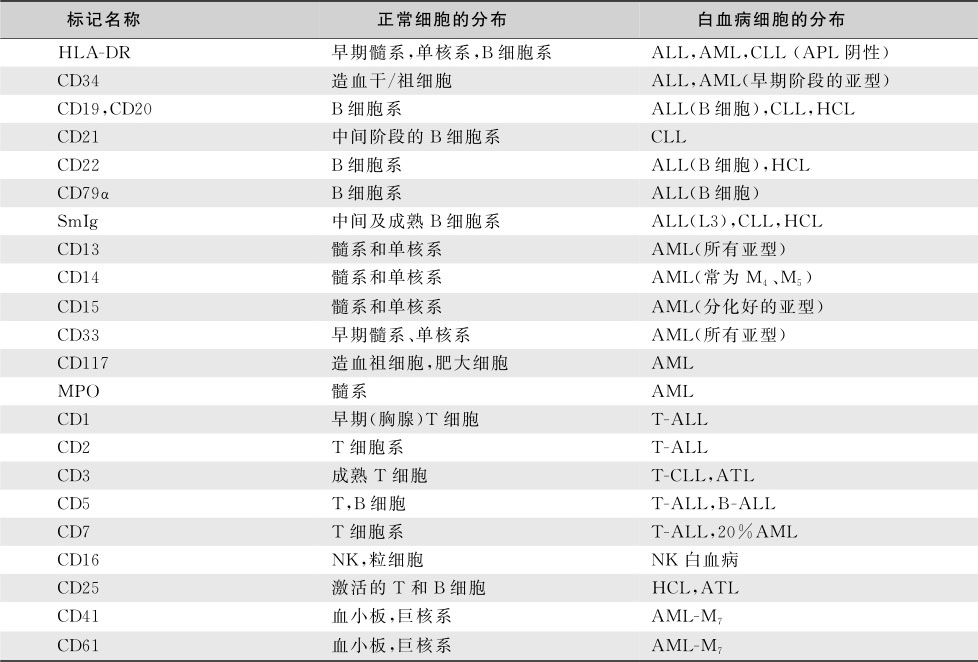
应用单克隆抗体(McAb)进行免疫分型过程中,有认为B系McAb中的CD10、CD19、CD22的特异性较好,T系McAb中的CD3、CD4、CD8的特异性较好,但表达率低,髓系McAb中的阳性表达率依次为CD33>CD13>CD14>CD15。60%ALL表达普通型 ALL抗原(CALLA,即CD10),CALLA为糖蛋白,偶见于正常早期淋巴细胞和其他非造血组织,CALLA阳性的ALL实际上是极早期B细胞。10%~20%的成人和5%~10%的儿童ALL有髓系抗原的表达(CD13和CD33),称表达髓系抗原的ALL(My + ALL);约20%~30%的AML表达淋系抗原,常见TdT、CD7、CD2和 CD19,称表达淋系抗原的 AML(Ly + AML)。诊断急性双系列(或双表型)白血病,WHO髓系肿瘤分类提出应根据欧洲白血病免疫分型研究组(EGIL)提出的积分系统(表16-3-5)计算积分,髓系积分>2分,淋系积分>2分才能确立。
表16-3-5 欧洲白血病免疫分类积分系统(EGIL)
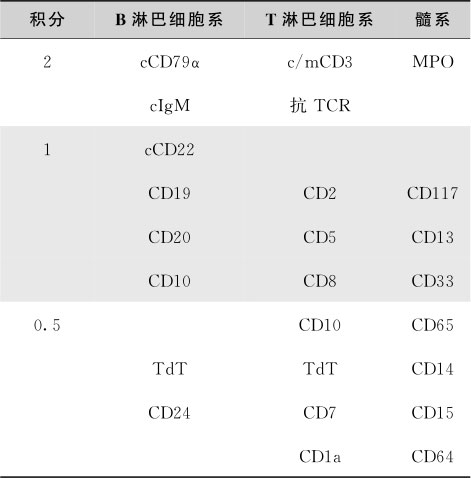
注:CD79α在某些前体T细胞白血病/淋巴瘤也有表达
(五)细胞遗传学检查
ALL约66%有特异性染色体变化,在有染色体畸变的AML中约60%有特异性染色体变化,因此骨髓细胞遗传学检查已成为急性白血病的形态学、免疫学、细胞遗传学和分子生物学(MICM)分类诊断的重要项目之一。AML的特异性染色体变化有:①t(8;21)(q22;q22):与急粒 M 2 型有特殊联系,据报道30%的 M 2 患者有t(8;21),t(8;21)往往伴有性染色体缺失,85%的男性患者缺少Y染色体,60%女性患者缺少X染色体。②t(15;17)(q22;q21):此易位限于急性早幼粒白血病(M 3 型),至少见于90%的M 3 患者;t(15;17)的检出对细颗粒和微颗粒型急性早幼粒白血病有重要价值,此外约1/3患者伴有+8。③t/del(11)(q23):本组染色体异常呈异质性,易位中最多见的是t(9;11),其他尚有t(11;9)(q23;p13)、t(10;11)(p11-p15;q23)和t(11;17)(q23;q21~25),它们均可出现在AML患者,约50%为急单 M 5a ,但也可见于T细胞 ALL。④inv/del(16)(q22):多见于急粒单白血病M 4 E 0 型。⑤t(9;22)(q34;q11):急粒白血病少见 Ph染色体异常,主要见于 M 1 型,它与慢粒不同,Ph(+)的AML初诊时多数细胞为正常二倍体。⑥t(6;9)(p21~22;q34):多见于M 2 或M 4 患者,极易涉及骨髓嗜碱性粒细胞但非绝对,约20%患者有 MDS病史。⑦inv(3)(q21;q26):可见于M 1 、M 2 、M 4 、M 7 和 MDS转变的AML白血病,伴血小板数升高,其他染色体异常如插入、易位等多见于 M 1 。⑧t(8;16)(p11;p13):系伴吞噬细胞增多,有吞噬红细胞现象的 M 5b 具有此异常。⑨t/del(12)(p11~13):可见于 AMLM 2 和M 4 ,其部分细胞向嗜碱性粒细胞分化。⑩+4:多见于M 4 或M 2 型AML。成人ALL 15%~20%有Ph染色体,其断裂点精确位置可能与慢粒不同,伴有Ph染色体的ALL常为非T非B型,有时为前B细胞型;t(4;11)最常见于新生儿 ALL,t(8;14)可见于 ALL L 3 型,t(1;19)见于前B细胞ALL;约20%ALL有染色体数量的增加,可达50~60条,这种超二倍体白血病化疗效果好。
(六)分子生物学检查
急性白血病分子水平的异常与疾病的发生、发展以及预后判断有密切关系。传统的细胞形态学和免疫学以及细胞遗传学检查已经不能满足急性白血病精准治疗的新理念,WHO(2001)造血与淋巴系统肿瘤分类标准已将基因异常作为最重要的确定疾病实体的依据之一。2008年修订颁布的WHO第4版分类标准在吸取最新研究成果的基础上,进一步推进了这一发展趋势。近年随着分子信号通路研究的逐步深入和靶向治疗药物的不断问世,基因分子水平异常检测不仅常规应用于急性白血病的诊断分类和预后判断,而且还成为疾病疗效评估和复发监测的一项重要手段。
分子水平检测急性白血病基因异常主要方法有FISH、PCR、RT-PCR、RQ-PCR以及高通量测序技术等。FISH可检测分裂中期和间期的细胞,克服了常规细胞遗传学检查细胞必须处于分裂中期的障碍。其缺点是灵敏度不及PCR方法。巢式RT-PCR和RQ-PCR技术是目前急性白血病临床疗效检测最为敏感的技术,并由此引入了“分子完全缓解(molecucular complete response)”的新概念。第二代测序技术(next generation sequencing,NGS)主要分为DNA-seq、RNA-seq和ChIP-seq等3类,对于个体化评估白血病克隆演变、药物靶点、DNA甲基化以及药物毒副作用等更加精准,但目前仅限于临床科研工作。第三代测序技术(next next generation sequencing)是通过合成互补链技术对数百万个DNA片段进行测序,克服了第二代测序技术依赖PCR扩增的信号放大技术,是真正意义的单分子测序,有望在21世纪上叶对白血病在内的血液肿瘤诊断与治疗带来突破性进展。
表16-3-6和表16-3-7分别为最常用于AML和ALL诊断分型的融合基因。
表16-3-6 AML常见的融合基因与染色体异常及白血病FAB类型的关系
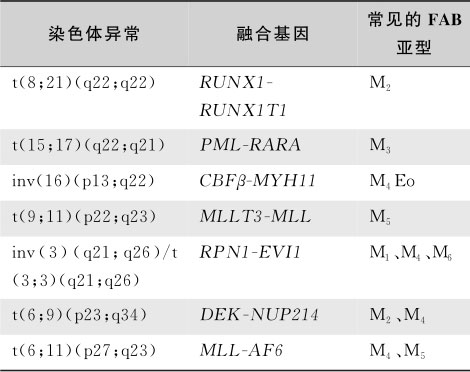
表16-3-7 ALL常见的融合基因与染色体异常及白血病类型的关系

三、诊断和鉴别诊断
(一)诊断
急性白血病时白细胞常显著增高,外周血液有数量较多的异常原始及幼稚细胞,但对白细胞不增多性白血病则必须借助骨髓检查才能发现白血病细胞。在未进行骨髓象检查之前,某些临床表现常易造成误诊。如儿童急性白血病常因发热、关节肿痛、心动过速而误诊为风湿热;有全血细胞减少的临床表现易误诊为再生障碍性贫血;某些急性白血病初起时可单系血细胞减少,如以粒细胞减少或血小板减少为首起表现的急性白血病常易误诊为粒细胞缺乏症或免疫性血小板减少症。上述情况只要及时进行骨髓象检查即可明确诊断。
ALL须注意和病毒相关的感染性单个核细胞增多症(infectious mononucleosis)鉴别。病毒相关的感染性单个核细胞增多症可有发热、皮疹、关节疼痛及淋巴结和肝脾大,外周血液和骨髓象中出现大量不典型淋巴细胞,易误诊为ALL,但病毒相关的感染性单个核细胞增多症贫血和血小板减少常不明显,病毒血清学和抗体检测有助于鉴别。儿童的神经母细胞瘤和横纹肌肉瘤及青少年和成年人的Ewing肉瘤及小细胞肺癌,有骨髓浸润时呈小圆细胞形态,如不注意时易误诊为ALL,肿瘤细胞的免疫表型和基因重排的类型有助于鉴别。药物引起粒细胞缺乏症的恢复期,骨髓可有早幼粒细胞显著增多,须注意和急粒相鉴别,前者常无贫血和血小板减少,且早幼粒细胞形态正常,存在环核浅染带,无Auer小体。粒细胞类白血病反应白细胞可超过50×10 9 /L且有核象左移,须注意与急粒鉴别,类白血病反应的骨髓象原粒细胞极少超过2%且NAP积分增高。低增生性急性白血病和急性再生障碍性贫血的鉴别,只要仔细检查骨髓并不困难,因为前者原始细胞百分比已达诊断急性白血病的标准。
(二)分型诊断
1.FAB分类标准
英美法协作组(FAB协作组)于1976年和1985年先后提出急性白血病的形态学诊断标准及修改建议,1991年又增补了AML的一项亚型,即AML微分化型(M 0 )。M 0 不能用通常的形态学和细胞化学方法找到肯定的髓系分化证据,但原始细胞可以通过单克隆抗体免疫标记和(或)超微结构(包括超微细胞化学)证实有髓系性质。M 0 的诊断标准为:骨髓原始细胞Ⅰ型+Ⅱ型在非红系(non-erythroid cell,NEC)中≥90%,原始细胞形态大多数类似ALL-L 2 的原始淋巴细胞、AML-M 1 原始细胞或少部分似AML-M 5 原始单核细胞,无嗜天青颗粒及Auer小体,常规细胞化学染色阴性。免疫表型无特异性高的淋系标志如cCD3、cCD79a和cCD22,但可表达特异性较低的淋系相关标志如CD2、CD4、CD7、CD10和CD19等,髓系分化抗原 CD13、CD14、CD33、CD64、CD65或 CD117等阳性,单抗检测细胞质髓过氧化酶(cMPO)阳性。急性未分化型白 血病 (acute undifferentiated leukemia,AUL)与AML-M 0 不同,AUL是指细胞表面无系列特异或系列相关抗原表达,细胞形态和细胞化学特征也无法确定哪一系列的白血病。有认为是否属于真正的AUL尚须经过基因分型的检测如髓过氧化物酶(MPO)基因表达、免疫球蛋白重链(IgH)或T细胞受体(TCR)基因重排等,证实无任何基因型和免疫学标志,才属于真正的AUL。
1986年天津白血病分类、分型讨论会提出了国内急性白血病的诊断标准。该标准系在FAB分类基础上提出。国内分型与FAB分型的不同之处是将AML-M 2 进一步区分为M 2a 和 M 2b 两个亚型(表16-3-8)。
表16-3-8 急性白血病国内诊断标准

注:①NEC指非红系细胞计数;②原粒细胞Ⅰ型指典型原粒细胞,胞质中无颗粒,Ⅱ型指有原粒细胞特征,胞质量少,有少量细小颗粒,原单核细胞Ⅰ型、Ⅱ型标准与原粒细胞类似
2.世界卫生组织分类标准
按WHO分类标准,在细胞形态学方面,不再将骨髓原始细胞区分为Ⅰ、Ⅱ两型;诊断AML骨髓原始细胞的标准从≥30%下降至≥20%。AML伴t(8;21)(q22;q22)、inv(16)(p13.1q22)/t(16;16)(p13.1;q22)及t(15;17)(q22;q12),不管原始细胞数量均可诊断相应类型 AML。而伴t(9;11)(p22;q23)、t(6;9)(p23;q34)、inv(3)(q21q26.2)/t(3;3)(q21;q26.2)及t(1;22)(p13;q13),当原始细胞<20%是否可诊断为 AML目前尚未有定论。ALL与LBL的区别以骨髓淋巴细胞25%为界限。急性白血病原始细胞表达两种以上系列特异性抗原而又不能确定为哪一系白血病称急性未定系列白血病(acute leukemia of ambiguous lineage,ALAL)。WHO(2008)分类标准将ALAL分为急性未分化白血病(acute undifferentiated leukemia,AUL)和混合表型急性白血病(mixed phenotype acute leukemia,MPAL)两类,后者根据细胞分子遗传学、免疫学和形态学检测结果,又可分为若干亚型。
3.ALL的免疫分型标准
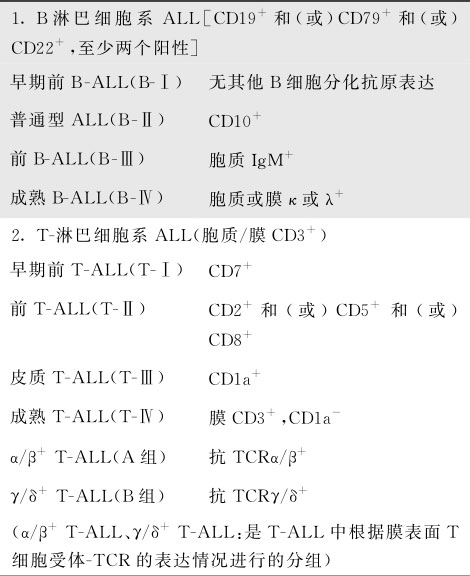
1994年在法国召开了欧洲白血病免疫学分型协作组(EGIL)会议,提出ALL四型21类法,即先按T、B淋巴细胞系和髓系抗原积分系统确定不同抗原积分,再按积分和抗原表达及分化程度把ALL分为四型(裸型、纯型、变异型、多表型)21亚型。由于该分型法比较复杂,不便临床医生记忆,国内学者卞寿庚等将其简化归纳为表16-3-9。
表16-3-9 ALL的免疫学分型(EGIL,1994)
续表

4.急性白血病的预后分型
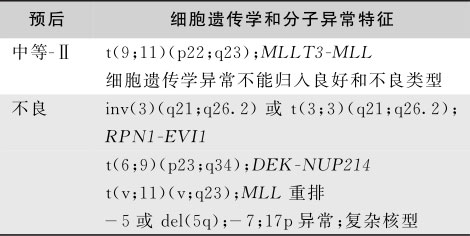
急性白血病患者的预后与发病时的年龄、白细胞计数、髓外浸润状态及FAB分型等多种因素有关,但在众多的预后相关因素中,白血病细胞的细胞遗传学和分子生物学特征与预后的关系最为密切。表16-3-10为2016年第2版美国NCCN(National Comprehensive Cancer Network)肿瘤临床实践指南推荐的根据细胞遗传学和分子异常特征预后分型。欧洲白血病网(LeukemiaNet)2012年AML的细胞遗传学和分子预后分型见表16-3-11。
表16-3-10 美国NCCN肿瘤临床实践指南推荐的AML的预后分型

表16-3-11 欧洲白血病网的AML预后分型

续表
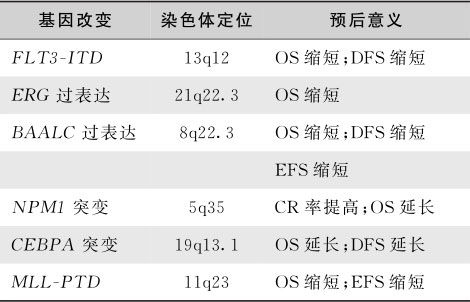
AML有40%~50%的患者骨髓细胞染色体检查为正常核型,称为细胞遗传学正常的急性髓细胞白血病(CNAML)。CN-AML的整体预后为中等,但近年随着分子生物学研究的进展,发现CN-AML患者可根据不同的基因改变作进一步预后分组(表16-3-12)。
表16-3-12 CN-AML的分子生物学预后分组
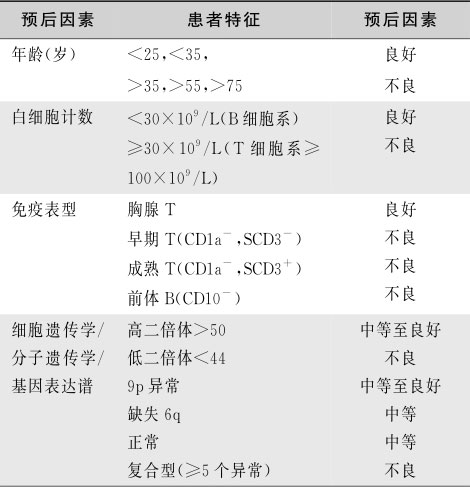
成人ALL与预后相关因素较AML更为复杂,发病年龄、初治时白细胞计数、免疫表型、细胞分子遗传学特征以前4周的治疗反应都与预后相关,详见表16-3-13。
表16-3-13 成人急性淋巴细胞白血病的预后因素
续表

四、治 疗
(一)治疗原则
急性白血病的治疗目标是彻底清除体内的白血病细胞,同时使正常造血功能得以恢复。化疗是实现这一目标的最主要手段,但目前常用的化疗药物,除糖皮质激素外几乎都有抑制造血功能的不良反应,并且对心、肝、肾、胃肠道也有毒副作用,所以急性白血病化疗宜采取循证医学基础上充分个体化的原则,根据白血病类型、病情程度以及患者重要脏器功能状态等客观条件灵活掌握。用药期间应严密观察,必要时调整剂量。同时必须加强支持治疗,防治感染和出血,以保证化疗的顺利进行。
(二)支持疗法
1.控制感染
复旦大学附属华山医院抗生素研究所及血液学研究室长期研究显示,20世纪80年代革兰阴性杆菌特别是铜绿假单胞菌感染一直是化疗后粒缺患者感染的主要病原体,但近年肺炎克雷伯菌和嗜麦芽假单胞菌、不动杆菌等的感染有所增加。随着第三代头孢菌素的广泛应用,白血病患者的细菌感染出现新的特点:①革兰阳性球菌逐步呈上升趋势,其中主要是凝固酶阴性的葡萄球菌和金黄色葡萄球菌,肠球菌、草绿色链球菌感染也有所增多。②致病菌出现耐药趋势,特别是产新型耐药酶如超广谱β-内酰胺酶(ESBLs)的细菌和新出现的耐药菌株感染明显增加。对怀疑感染发热患者应反复寻找病原菌并进行药敏试验。在细菌培养有结果前先按经验早期应用广谱高效抗生素,以后再根据病原学检查及药敏试验结果调整用药。
对产ESBL细菌的治疗可参考以下原则:①如怀疑产ESBL菌感染时,不管体外药敏结果是否敏感,应避免使用青霉素类、头孢菌素类抗生素;②选择使用碳青霉烯类抗生素、加β-内酰胺酶抑制剂抗生素(头孢哌酮/舒巴坦、头孢哌酮/他唑巴坦、哌拉西林/他唑巴坦、替卡西林/克拉维酸及等)、氨基糖苷类及头霉素类抗生素。嗜麦芽窄食单胞菌感染在插管和置管患者亦较常见,该菌对亚胺培南耐药,应选择含β-内酰胺酶抑制剂抗生素或头孢吡肟、环丙沙星、复方磺胺甲基异
 唑等。对于耐甲氧西林金黄色葡萄球菌(MRSA)和耐甲氧西林凝固酶阴性葡萄球菌(MRCNS)感染首选万古霉素或去甲万古霉素,肾功能有损害者可选择替考拉宁。利奈唑胺在肺组织液中浓度相对较高,但长期使用可能引起造血功能的抑制,可根据具体情况酌定。
唑等。对于耐甲氧西林金黄色葡萄球菌(MRSA)和耐甲氧西林凝固酶阴性葡萄球菌(MRCNS)感染首选万古霉素或去甲万古霉素,肾功能有损害者可选择替考拉宁。利奈唑胺在肺组织液中浓度相对较高,但长期使用可能引起造血功能的抑制,可根据具体情况酌定。
真菌感染如局限在口腔或咽部,可涂搽制霉菌素。深部真菌感染以念珠菌最常见,包括白念珠菌、热带念珠菌、光滑念珠菌、近平滑念珠菌、克柔念珠菌等。曲霉菌和隐球菌感染近来也不少见。常用的抗真菌药有三唑类(氟康唑、伊曲康唑、伏立康唑)、棘白霉素类(卡泊芬净、米卡芬净)、大环内酯多烯类(两性霉素B及两性霉素B脂质体)等。氟康唑对白念珠菌、近平滑念珠菌、热带念珠菌敏感,对新型隐球菌敏感率达89%。光滑念珠菌、克柔念珠菌耐药,对曲霉菌无效。伊曲康唑抗菌谱广,可治疗深部白念珠菌和曲霉感染,不宜用于尿路感染,肾功能减退,肌酐清除率<30ml/min禁用。伏立康唑为第二代三唑类抗真菌药,抗菌谱包括耐氟康唑和伊曲康唑的念珠菌属,新型隐球菌、毛孢子菌、球孢子菌、曲霉菌、组织胞浆菌。卡泊芬净作用于真菌细胞壁的葡聚糖合成酶,主要用于治疗对三唑类及两性霉素B耐药的曲霉菌和念珠菌属感染。两性霉素B可与真菌细胞膜上甾醇结合,使真菌细胞膜内重要物质外漏,致其死亡。主要用于治疗耐氟康唑和伊曲康唑的念珠菌属、曲霉菌、毛霉菌、球孢子菌、皮炎芽生菌、组织胞浆菌感染。两性霉素B不易透过血脑屏障,治疗隐球菌性脑膜炎需要和氟胞嘧啶合用。由于两性霉素B肾毒性显著,对于总量>0.5g无效或不能耐受者,深部真菌感染伴肾功能减退(血肌酐>221μmol/dl)者,可考虑用两性霉素B脂质体治疗。
急性白血病患者的病毒感染以单纯疱疹病毒(HSV)、水痘-带状疱疹病毒(VZV)和巨细胞病毒(MCV)感染为多见。无环鸟苷(阿昔洛韦)为病毒DNA多聚酶抑制剂,对HSV、VZV及CMV感染都有预防和治疗作用。更昔洛韦是目前有效的抗MCV药,但有导致粒细胞减少的副作用。阿糖腺苷亦可用于HSV、VZV感染的治疗,但对MCV感染无效。
由于急性白血病患者机体免疫功能低下,对严重细菌和病毒感染疗效不佳者可静脉滴注大剂量丙种球蛋白,每天约20g,共5天。
2.纠正贫血
纠正贫血最有效的方法是通过化疗有效杀灭白血病细胞,使骨髓正常造血功能得到恢复。化疗前和化疗期间如有显著贫血可酌量输注红细胞悬液。合并自身免疫溶血性贫血者可采用糖皮质激素治疗。如白血病获得缓解,但血红蛋白恢复不满意,应注意是否存在铁利用障碍,可酌情加用丙睾酮注射,司坦唑口服或红细胞生成素皮下注射。
3.防治出血
白血病获得缓解是纠正出血的最有效方法。血小板计数<2×10 9 /L伴出血可输注单采血小板。急性白血病并发弥散性血管内凝血,一经肯定诊断,应迅速给予低分子肝素治疗,持续至凝血现象好转。当弥散性血管内凝血并发纤维蛋白溶解症,可在肝素治疗同时并用抗纤溶药物(如对羧基苄胺、氨甲环酸等)。局部出血(如鼻咽部)用填塞或明胶海绵止血。
4.纠正高尿酸血症
大量白血病细胞破坏分解时血尿酸增高,有时尿路为尿酸结石梗阻,引起少尿等急性肾衰竭。别嘌醇为黄嘌呤氧化酶抑制剂,能阻断次黄嘌呤和黄嘌呤变为尿酸,可纠正尿酸过高。剂量为10mg/kg体重,每天3次口服,共5~6天。当血尿酸超过595μmol/L,应大量输液和碱化尿液。
(三)化学治疗
应先确定白血病类型,再选择适当化疗方案。为了防止耐药性产生,初治患者应选用对白血病细胞敏感的药物,在患者可耐受情况下尽可能加大剂量,采用联合或序贯化疗,争取在短时间内(2~3周或1~2疗程)杀伤大量肿瘤细胞而使疾病进入缓解期。化疗疗程以超过白血病细胞增殖周期或倍增时间为妥。急性白血病细胞的倍增时间为4~5天,所以抗白血病药物应连续应用5~10天,使进入周期的所有细胞都受到药物作用。为了避免造血系统不可逆性损害,应该间歇用药,以使正常血细胞得以恢复。正常血细胞复原较白血病细胞为快,而血细胞从骨髓增殖池释放至外周血中大约需10~15天,因而间歇期应以2周左右为好。这样既能杀灭大量白血病细胞,又有利于血象恢复。
急性白血病化疗可分成诱导缓解和缓解后继续治疗两大阶段:①诱导缓解治疗:目标是应用化疗药物短期内将白血病细胞减少到一定程度,正常造血功能得以恢复,患者症状消失,一般检查方法血片中不能找到白血病细胞。要特别重视初治疗效,力争1~2个疗程即达到完全缓解(complete remission,CR)。对全血细胞减少伴骨髓增生低下的老年急性白血病患者,如全身情况较差,也可先用小剂量化疗,如阿糖胞苷、阿克拉霉素或(高)三尖杉碱,待血象稍见上升,再按常规剂量化疗方案治疗。对此类患者必须反复检查骨髓,随时调整剂量。②缓解后治疗:急性白血病患者经治疗获得CR后,体内仍残留一定数量白血病细胞,必须继续应用抗白血病药物,以消灭尽可能多的残留白血病细胞,从而达到长期无病生存乃至彻底治愈的目标。缓解后继续治疗期药物要求耐药性出现缓慢,且与诱导缓解药物无交叉耐药性。对继续治疗时间目前尚无统一意见,大多主张AML在完全缓解后巩固强化6~8个月即停药;ALL患者经巩固治疗后,尚须维持治疗3年之久。
急性白血病CR标准:①形态学无白血病状态:骨髓白血病细胞<5%;外周血无幼稚细胞;髓外无浸润病变。②造血正常:骨髓三系增生;外周血中性粒细胞绝对值>1.0×10 9 /L;血小板计数>100×10 9 /L;不依赖输血。③细胞遗传学完全反应:以前如发现有细胞遗传学异常,现恢复正常。④分子生物学完全反应:分子检测转阴(目前主要对APL和Ph + 白血病患者而言)。评估时还需要注意:同时达到①和②者,可以称之为形态学CR。如果患者其他各项指标均符合CR,但血小板计数和(或)中性粒细胞绝对值不能完全恢复,则称之为CRi(CR with incomplete blood count recovery)。
1.AML(非APL)的治疗
(1)诱导缓解治疗:
蒽环类药物柔红霉素与阿糖胞苷联合的DA方案是除APL以外其他各型AML最常用的诱导治疗方案(表16-3-14)。完全缓解率为60%~85%,但对于60岁以上的患者CR率只有45%~55%。由于柔红霉素对心脏具有明显毒性,因此一般应限制累积剂量不超过550mg/m 2 ,老年患者及原有心脏疾病患者更需谨慎使用。对原有冠状动脉疾病或充血性心力衰竭史者,发生心脏毒性的危险性更高,可给予右雷佐生(dexrazoxane)以减少风险。关于诱导治疗中柔红霉素的最合适剂量,国外两个随机研究报告,与阿糖胞苷联合应用时,柔红霉素90mg/(m 2 ·d)连续3天与45mg/(m 2 ·d)连续3天比较,前者的完全缓解率更高,但对支持治疗的要求也更高。去甲氧柔红霉素是柔红霉素的衍生物,其特点是细胞毒作用较柔红霉素更强,对中枢神经系统白血病有更好的预防和治疗作用,心脏毒性较低,并且与其他蒽环类药物无交叉耐药性。一些临床研究显示,应用去甲氧柔红霉素代替DA方案中的柔红霉素,疗效更优。此外,蒽醌类药物米托蒽醌与阿糖胞苷组成方案也可用于AML的诱导缓解治疗。20世纪80年代国外有作者报告在DA方案的基础上加依托泊苷(VP16)对<55岁的年轻患者能进一步提高完全缓解率,延长生存期,尤其对于 M 4 和 M 5 型患者。但这一结果并未得其他研究者的广泛认同。三尖杉酯碱或高三尖杉酯碱与阿糖胞苷组成的HA方案是国内常用于AML的诱导缓解治疗的另一方案,其CR率为76.0%,与DA方案相似。但应注意三尖杉酯碱或高三尖杉酯碱也有较强的心脏毒副作用。1995年中国医学科学院血液学研究所设计以高三尖杉酯碱与阿糖胞苷+柔红霉素组成的HAD方案治疗成人初治AML取得85%的完全缓解率,其中一疗程完全缓解率达80%。据认为HAD方案的优势主要在于高三尖杉酯碱与柔红霉素之间存在一定的协同作用。
表16-3-14 成人急性白血病诱导缓解治疗的常用化疗方案

上述各药物简称的全名如下:VCR:长春新碱;P:泼尼松;DAUN:柔红霉素;ADM:多柔比星;CTX:环磷酰胺;L-ASP:门冬酰胺酶;DXM:地塞米松;H:三尖杉碱(或高三尖杉酯碱);Ara-C:阿糖胞苷;IDA:去甲氧柔红霉素;VP16:依托泊苷;MTX:甲氨蝶呤;Ara-C:阿糖胞苷
大剂量Ara-C(HD-Ara-C)在AML的疗效已得到国外多项研究的肯定。但用于诱导缓解治疗因治疗相关死亡率相对较高。目前除年轻患者外,多将大剂量Ara-C用于完全缓解后的治疗。
2016年第2版美国NCCN(National Comprehensive Cancer Network)肿瘤临床实践指南建议AML(非APL)治疗按患者年龄60岁为界,分为两组。<60岁组患者的主要推荐方案:①阿糖胞苷100~200mg/(m 2 ·d),持续静脉输注×7天,去甲氧柔红霉素12mg/(m 2 ·d)(或柔红霉素60~90mg/m 2 )×3天;②阿糖胞苷200mg/(m 2 ·d),持续静脉输注×7天,柔红霉素60mg/m 2 ×3天,克拉屈滨5mg/m 2 ×5天。如年龄≤45岁,可考虑选用更加强烈的方案:①大剂量阿糖胞苷2g/m 2 ,每12小时1次,共6天(或3g/m 2 ,每12小时1次,共4天),去甲氧柔红霉素每天12mg/m 2 (或柔红霉素60~90mg/m 2 )×3天。②氟达拉滨30mg/(m 2 ·d),第2~6天,阿糖胞苷2g/(m 2 ·d),在氟达拉滨注射后4小时开始滴注,维持4小时以上。去甲氧柔红霉素8mg/(m 2 ·d),第4~6天,G-CSF每天皮下注射,第1~7天。
年龄≥60岁患者可酌情选择的治疗策略为:①标准方案:阿糖胞苷每天100~200mg/m 2 ,持续静脉输注×7天,去甲氧柔红霉素12mg/(m 2 ·d)(或柔红霉素45~90mg/m 2 ,或米托蒽醌12mg/m 2 )×3天;②低强度治疗(皮下注射阿糖胞苷、阿扎胞苷、地西他滨)。③临床试验。④最好的支持治疗(羟基脲、输血等)。一般来说,de novo AML(非继发于其他造血系统疾病或治疗相关AML)患者如无不良细胞遗传学或分子标志可选择标准方案或低强度治疗。如有不良细胞遗传学/分子标志者,多考虑选用低强度治疗或临床试验。但标准方案也非绝对禁忌。
(2)缓解后治疗:
多数研究者认为诱导完全缓解后的治疗方案和强度直接影响患者的长期生存率。美国东部肿瘤协作组(Eastern Cooperative Oncology Group,ECOG)比较以下4个治疗组的远期疗效:①停药观察;②长期小剂量维持治疗;③常规剂量联合化疗巩固加长期小剂量维持化疗;④含HD-Ara-C联合方案巩固治疗后停药,不再维持治疗。4组的4年DFS率依次为0%、15%、20%和30%。
HD和中剂量(ID)Ara-C单用或联合蒽环类、鬼臼类等药物是当前广泛使用的完全缓解后的强化巩固治疗方案。美国癌症与白血病协作组(CALGB)的研究显示,接受标准剂量阿糖胞苷+柔红霉素诱导治疗以及3个疗程HDAra-C巩固治疗的患者4年DFS达44%。治疗相关死亡率为5%,严重神经毒性反应发生率为12%。如果再按细胞遗传学危险度分层后进行比较,具有良好细胞遗传学改变患者的DFS为60%,中危患者为30%,不良预后者为12%。但注意到HD-Ara-C对有MDS病史及老年患者的疗效并不理想。
2016年第2版美国NCCN肿瘤临床实践指南建议AML(非APL)<60岁患者、如预后分型良好或中等,可给予HD-Ara-C 3g/m 2 ,静脉输注3小时以上,每12小时1次,第1天、第3天、第5天,共3~4疗程。预后中等患者也可考虑异基因造血干细胞移植。患者预后分型不良者,应首选异基因造血干细胞移植。年龄≥60岁的患者,巩固治疗可选择:①标准剂量的阿糖胞苷联合蒽环类药物(去甲氧柔红霉素、柔红霉素或米托蒽醌)。②含ID-Ara-C(1.0~1.5g/m 2 )的方案。③如符合条件,也可行减低剂量的异基因造血干细胞移植。
2.急性早幼粒细胞白血病的治疗
(1)诱导缓解治疗:
急性早幼粒细胞白血病(acute promyelocytic leukemia,APL)因15号与17号染色体之间易位形成 PML / RARA 融合基因,其表达的PML/RARA融合蛋白通过阻断细胞分化和凋亡导致APL发生。采用全反式维甲酸(ATRA)诱导分化是目前国际上公认的伴t(15;17)急性早幼粒细胞白血病的首选诱导缓解方案。ATRA可与维甲酸受体结合,加快PML/RARA融合蛋白的降解,使早幼粒细胞继续分化成熟。常用剂量为45mg/(m 2 ·d),或60~80mg/d连续口服至CR。1991年华东地区全反式维甲酸协作组会议共总结787例急性早幼粒细胞白血病,初治603例,完全缓解率为85.4%,复治60例,完全缓解率为74%;单独应用ATRA治疗组,其完全缓解率为85.2%。1995年上海第二医科大学附属瑞金医院又报道以30~40mg/d的剂量治疗,同样可以达到87.5%的完全缓解率。ATRA治疗伴t(15;17)APL一般数天内即可纠正患者合并的凝血功能障碍,但可出现白细胞增多引起的维甲酸综合征(近年又称APL分化综合征)、颅内压增高、皮肤黏膜干燥、消化道反应、肝功能损害、外阴水肿甚至溃疡等不良反应。其中以维甲酸综合征最为严重,发生率约为20%~25%。主要临床表现为发热、肺间质浸润、胸腔积液、呼吸窘迫甚至呼吸衰竭。可伴有或不伴有白细胞计数增高。紧急救治方法为地塞米松20mg/d静脉注射,连续3天,并正压持续吸氧等各种对症处理。ATRA的另一缺点是不能用作维持治疗。诱导缓解成功后,如不加用其他治疗措施,3~4个月后大多复发。
继ATRA之后我国学者又首创砷剂治疗APL取得成功。最常用的砷剂有三氧化二砷(亚砷酸,As 2 O 3 )、硫化砷(As 2 S 3 )和四硫化四砷(As 4 S 4 ),其中以 As 2 O 3 应用最广。砷剂治疗APL的主要机制为诱导早幼粒白血病细胞凋亡。亚砷酸的常规用法是5mg/m 2 或10mg/m 2 ,加入5%葡萄糖溶液500ml中静脉滴注3~4小时,连续28天为1疗程。间歇1~2周,再重复1个疗程,连用2个疗程未缓解可视为无效。CR率90%~98%,并可较早获得分子完全缓解。砷剂的另一重要特点是与ATRA无交叉耐药。ATRA治疗后复发和难治的患者应用As 2 O 3 再诱导治疗,CR率为78%~93%。砷剂的主要毒副作用有白细胞增高、APL分化综合征、心电图Q-T间期延长、周围神经病变、皮疹及胃肠道反应等。近年国内外学者尝试ATRA联合三氧化二砷用于初治APL患者的诱导治疗。结果表明ATRA联合As 2 O 3 诱导缓解要比单用ATRA或As 2 O 3 达到CR时骨髓细胞PML-RARA转录本更低,因此复发率亦更低。
尽管以ATRA为基础的治疗使APL的预后大为改观,但仍有部分患者存在复发的风险。西班牙和意大利协作组通过对217例APL患者的随访观察表明,患者初诊时外周血白细胞和血小板计数是预后的独立因素。白细胞计数≤10×10 9 /L,血小板计数>40×10 9 /L属低危组(24%);白细胞计数≤10×10 9 /L,血小板计数≤40×10 9 /L为中危组(53%);白细胞计数>10×10 9 /L则归入高危组(23%)。
较长时期内,Ara-C在APL诱导缓解和缓解后治疗中的的作用一直不甚明了。但2008年国外临床试验的结果显示对于初治时白细胞数≥10×10 9 /L、血小板数<4×10 9 /L的患者采用Ara-C与DNR联合诱导和巩固治疗,完全缓解率和3年存活均较不含Ara-C的对照组具有一定的优势。
2016年第2版NCCN指南建议初治高危APL患者(WBC>10×10 9 /L)如能耐受蒽环类药物者,可选择以下治疗方案:①每天给予ATRA45mg/m 2 ,分次口服,直至临床缓解,然后加用柔红霉素每天50mg/m 2 ×4d(或60mg/m 2 ×3d),阿糖胞苷每天200mg/m 2 ×7d;或②每天ATRA 45mg/m 2 ,分次口服,d1~36,根据年龄调整剂量去甲氧柔红霉素6~12mg/m 2 ,d2、d4、d6、d8,亚砷酸每天0.15mg/kg静脉输注,d9~36。③每天给予ATRA45mg/m 2 ,分次口服,直至临床缓解,去甲氧柔红霉素12mg/m 2 ,d2、d4、d6、d8;④如患者不能耐受蒽环类药物,可每天给予ATRA 45mg/(m 2 ·d),分2次口服,亚砷酸每天0.15mg/kg静脉输注。直至骨髓缓解。
低中危APL患者(WBC<10×10 9 /L)可选择以下治疗方案:①ATRA每天45mg/m 2 ,分次口服,直至临床缓解,亚砷酸每天0.15mg/kg静脉输注,直至骨髓缓解。②ATRA每天45mg/m 2 ,分次口服,直至临床缓解,加用柔红霉素每天50mg/m 2 ×4d(或每天60mg/m 2 ×3d),阿糖胞苷每天200mg/m 2 ,×7d;③ 每天ATRA 45mg/m 2 ,分次口服,直至临床缓解,去甲氧柔红霉素每天12mg/m 2 ,d2、d4、d6、d8。
(2)缓解后治疗:
高、中、低危患者均可采用维甲酸+亚砷酸±蒽环类的方案进行巩固治疗。如单纯采用蒽环类+阿糖胞苷进行巩固治疗,阿糖胞苷的剂量宜适当加大,建议每天1~2g/m 2 ×(4~5)d。PCR监测分子生物学阴性结果需要2年,以便及时发现分子生物学水平的复发。
目前多数学者主张首次获得CR的APL患者,不推荐立即行造血干细胞移植。造血干细胞移植的时机一般可选择在CR2期。欧洲血液和骨髓移植组织报道,APL患者CR2期行异基因造血干细胞移植的总生存率、无病生存率、复发率及治疗相关死亡率分别为58%、57%、15%和33%;自体造血干细胞移植为40%、45%、44%和25%。故无合适供体者采用自体造血干细胞移植亦不失为一项有效治疗措施。
ATRA的应用使APL患者生存期显著延长,但中枢神经系统白血病的发生率也随之多见,尤其是高危患者。应将中枢神经系统白血病的预防作为APL患者缓解后治疗的一项常规措施。
3.ALL的治疗
(1)诱导缓解治疗:
泼尼松与长春新碱联合的VP方案,可使标危儿童ALL的完全缓解率达95%。但该方案用于成人ALL的诱导缓解治疗,CR率仅为47%,在VP加用蒽环类药物,其CR率可提高到83%。目前由VP方案加柔红霉素组成的VDP方案已普遍成为ALL诱导缓解治疗的常用基础方案。在VDP方案中蒽环类的剂量和用法一些学者也进行过研究。去甲氧柔红霉素每天12mg/m 2 ,2~4天诱导治疗ALL的死亡率高达50%,而减低剂量至每天10mg/m 2 ,2~3天,其相关死亡率降至9%。柔红霉素或米托蒽醌持续静滴并不优于静脉推注,而且柔红霉素用药延长至1周也不优于3天的疗效。
地塞米松与泼尼松比较,用于ALL的治疗主要有两方面的优势,①抗白血病作用更强,体外实验证明地塞米松对ALL细胞的作用较泼尼松强16倍。②更容易渗透进入中枢神经系统,在脑脊液中药物浓度更高,半衰期更长。荷兰的一项历史对照研究显示,地塞米松与泼尼松比较,ALL患者3年EFS分别为80%与66%。另一些临床试验也证实,在减少ALL中枢神经系统白血病的复发率及3年EFS方面,地塞米松优于泼尼松。门冬酰胺酶(L-ASP)是另一种常用于ALL诱导缓解的药物,在VDP方案中加入门冬酰胺酶的VDLP方案也是目前常用的ALL诱导治疗方案。国外有临床研究显示,门冬酰胺酶并不能提高诱导治疗的CR率,但可延长缓解期。一些非随机研究认为,在VDLP方案基础上加入环磷酰胺(VDCP-L)可进一步提高CR率,尤其适用于成人T-ALL患者。近年已有聚乙二醇(PEG)与门冬酰胺酶共价结合的制剂上市,其优点除了底物专一性高、过敏反应少外,体内半衰期也显著延长,使给药次数大为减少。
含hyper-CVAD方案的诱导缓解治疗是近年来推出的一种新的成人ALL治疗策略,与上述方案不同之处主要在于将环磷酰胺改为分段使用,并增加了交替使用大剂量阿糖胞苷(HD-Ara-C)和大剂量甲氨蝶呤(HD-MTX)。研究结果表明诱导缓解率和长期生存率较VAD(长春新碱、阿霉素、地塞米松)更高。
另有报道在含hyper-CVAD方案基础上加用抗CD20单抗美罗华治疗Burkitt 淋巴瘤/白血病,CR率为86%,3年OS、EFS、DFS分别达89%、80%和88%。与单用含hyper-CVAD方案的历史对照组比较,优势较为明显。
Ph染色体阳性 ALL(Ph + ALL)占成人 ALL的20%~30%左右。随着年龄的增加,发生率也随之增高。在50岁以上的ALL患者中发生率可>40%。Ph + ALL主要见于前B-ALL,90%以上的患者表达CD34;50%以上的患者还表达髓系抗原标记,如CD13、CD33等。临床上白细胞计数常增高,但脾脏及淋巴结肿大少见。在酪氨酸激酶抑制剂(TKIs)问世之前,Ph + ALL的预后很差,化疗虽然能使60%以上的患者获得CR,但易复发,平均缓解期仅为9个月,其5年DFS低于10%~20%。来自GIMEMA临床试验的一组资料显示,101例Ph + 成人ALL患者中p190 BCR/ABL阳性占(59例)57.6%,p210 BCR/ABL 阳性占42.4%(p210 BCR/ABL单独阳性23例,p210与p190共同阳性19例)。均采用泼尼松、长春新碱、大剂量柔红霉素(总剂量达270mg/m 2 )和L-ASP诱导治疗,继以 HDAra-C联合米托蒽醌强化治疗,并在CR1期行异基因或自体造血干细胞移植。在可评估的92例资料中,治疗相关死亡率为15.2%,总CR率为67.4%,其中p190 BCR/ABL阳性组CR率分别为69.8%,p210 BCR/ABL阳性组为64.1%。两组间无显著性差异。52例行强烈再诱导治疗后进行造血干细胞移植,36例(20例异基因造血干细胞移植,16例自体造血干细胞移植)获得持续CR。作者评估时6/20、4/16例仍然处于持续缓解之中。未接受造血干细胞移植的16例无1例存活。研究还认为p190 BCR/ABL阳 性组在OS和DFS方面要优于p210 BCR/ABL阳性组。
甲磺酸伊马替尼(Imatinib Mesylate)在CML治疗取得成功以后,国外开展了治疗Ph + ALL的临床试验。Ⅰ期临床试验20例Ph + ALL异基因造血干细胞移植后复发的病例,应用伊马替尼600mg/d治疗,有11例(55%)获得完全血液学缓解,4例骨髓完全缓解,但血常规未完全恢复,5例患者为难治性或仅获得部分缓解。在有效的病例中,应用伊马替尼治疗的前4周,骨髓或外周血供者嵌合体增加到96%,提示伊马替尼对Ph + 白血病细胞有选择性抑制作用,从而间接促进Ph - 细胞增殖。
美国MD Anderson肿瘤中心研究了在初发Ph + ALL患者中应用伊马替尼联合hyper-CVAD方案的疗效,8个疗程的诱导缓解和巩固治疗中,每疗程的第14天给予伊马替尼,8个疗程结束后给予伊马替尼600mg/d,维持治疗1年。结果显示这种联合治疗是安全的,并且缓解率较高。
随着TKIs联合化疗药物治疗成人ALL经验的逐步积累,2016年第1版美国NCCN肿瘤临床实践指南推荐将成人ALL的治疗分为Ph + ALL和Ph - ALL两大类,两类患者再根据年龄分为青少年及年轻成人(adolescent and young adult,AYA)组(15~39岁)和成人(adult)组(≥40岁)。
Ph + ALL AYA组患者诱导治疗可选择:①COG AALL-0031方案,即长春新碱、泼尼松(或地塞米松)、门冬酰胺酶±柔红霉素,该方案中的柔红霉素可以酌情不用。伊马替尼在巩固治疗阶段应用。②伊马替尼或达沙替尼联合hyper-CVAD方案。③伊马替尼联合VDCP方案。
Ph + ALL成人组患者如年龄<65岁,且不存在严重合并症,可参照Ph + ALL AYA组的诱导治疗策略,如选择TKI联合hyper-CVAD或VDCP方案。如患者伴有严重合并症或年龄≥65岁,可选择TKIs联合长春新碱+地塞米松方案,甚至TKIs仅与糖皮质激素合用。
Ph - ALL AYA组患者诱导治疗可选择①GRA ALL-2003方案(柔红霉素、长春新碱、泼尼松、环磷酰胺、门冬酰胺酶),或②COG AALL-0434方案(柔红霉素、长春新碱、泼尼松、门冬酰胺酶,T-ALL可在巩固治疗中加入奈拉滨),也可选用在VDLP(长春新碱、柔红霉素、门冬酰胺酶、泼尼松)基础上加用HD-MTX;对于CD20阳性患者,可考虑采用hyper-CVAD联合利妥昔单抗治疗。
Ph - ALL成人组患者诱导治疗可选择的主要方案有:①CALGB 8811 Larson方案(柔红霉素、长春新碱、泼尼松、门冬酰胺酶、环磷酰胺),如年龄≥60岁,环磷酰胺、柔红霉素以及泼尼松适当减量,也可不用环磷酰胺。②hyper-CVAD方案±利妥昔单抗。③MRC UKALLⅦ/ECOG2993方案:Ⅰ期诱导(柔红霉素、长春新碱、泼尼松、门冬酰胺酶),Ⅱ期诱导(环磷酰胺、阿糖胞苷、6-巯基嘌呤)。
(2)缓解后治疗:
成人ALL取得CR后必须进行巩固和维持治疗,时间应坚持2~3年,期间应密切监测微小残留病(minimal residual disease,MRD)状态。但Ph + ALL患者,首先需考虑异基因造血干细胞移植,移植后继续以TKIs维持治疗。如供体不能获得,应采用TKIs+多药联合方案巩固治疗。维持治疗前6个疗程的巩固治疗对于提高患者的长期无病存活率尤为重要。巩固治疗方案可选择诱导治疗推荐的方案交替进行,如VDLP、VDCP-L、hyper-CVAD等。每疗程之间一般间隔期为2~3周,不宜过长。维持治疗一般每周1次甲氨蝶呤+每天6-巯基嘌呤,每月1次长春新碱/泼尼松,Ph + ALL患者维持治疗需要联合TKI。
4.难治性急性白血病的治疗
(1)难治性急性白血病诊断标准:
德国AMLCG协作组提出的4项标准得到较为广泛的认可:①标准方案诱导治疗2个疗程不能缓解;②CR1后6个月内复发:③CR1后6个月后复发,且原诱导缓解方案再诱导治疗无效:④二次或多次复发。从中可以看出,所谓的难治性白血病其实包括原发性难治和复发两类患者。
难治性急性白血病的治疗策略,可参考以下原则:①选择与原治疗方案无交叉耐药性的药物组成新的治疗方案。②采用与常规药物作用机制不同的抗白血病新药。③将常规化疗药物加大剂量使用。④年龄较轻、一般状况尚可、早期复发患者,尽量予以积极治疗;高龄或一般情况较差、多次复发患者,可酌情采用较保守治疗措施。
(2)难治性AML的治疗:
一般认为,CR期超过12个月的复发患者较12个月内复发的患者疗效相对较好。化疗方案的选择原则是:①采用无交叉耐药的化疗药物;②HD-Ara-C与其他新药等联合应用。
氟达拉滨是一种合成的嘌呤类似物,其结构类似于Ara-C,在Ara-C的2位上加氟,增强了对腺苷脱氨酶的脱氨作用,在糖的部位增加了磷,则使其水溶性增强。在体内经磷酸化成为有活性的三磷酸形式F-Ara-ATP,通过抑制核糖核酸还原酶、DNA多聚酶、DNA引物酶、DNA连接酶的作用而抑制DNA的合成,并能部分抑制RNA聚合酶Ⅱ减少蛋白质的合成。由氟达拉滨、大剂量阿糖胞苷联合G-CSF组成的FLAG方案是目前常用的难治与复发AML的治疗方案。其特点是G-CSF可动员静止期白血病细胞进入增殖周期,氟达拉滨可增强阿糖胞苷的细胞毒作用。FLAG方案治疗难治复发白血病的CR率达50%~75%。对晚期复发(停药>6个月)患者的CR率明显好于早期复发(停药<6个月)和难治患者。
近年来,除氟达拉滨外,含其他嘌呤类似物如克拉屈滨(cladribine)、氯法拉滨(clofarabine)的化疗方案在难治/复发AML的临床试验中也取得鼓舞人心的疗效,缓解率达到30%~65%。以克拉屈滨为基础的化疗方案主要有两种,具体用法:①CLAG方案[克拉屈滨5mg/(m 2 ·d),d1~5;阿糖胞苷2g/(m 2 ·d),d1~5;G-CSF 300μg/d,d0~5]。②CLAM方案[在CLAG方案基础上加米托蒽醌10mg/(m 2 ·d),d1~3]。含氯法拉滨的代表性方案有:①氯法拉滨25mg/(m 2 ·d)×5d,阿糖胞苷每天2g/(m 2 ·d)×5d,联合G-CSF。②氯法拉滨22.5mg/(m 2 ·d)×5d,去甲氧柔红霉素6mg/(m 2 ·d)×3d,阿糖胞苷0.75g/(m 2 ·d)×5d,联合 G-CSF。③氯法拉滨22.5mg/(m 2 ·d)×5d,去甲氧柔红霉素10mg/(m 2 ·d)×3d。此外,去甲基化药物地西他滨(decitabiine)和阿扎胞苷(5-azacitidine)对部分难治/复发AML患者有效。甲苯磺酸索拉非尼(Sorafenib)是一种激酶抑制剂,在体外显示可抑制多种涉及肿瘤细胞内信号转导、血管生成和凋亡相关的激酶,可以和去甲基化药物联合试用于 FLT3-ITD 突变患者。
拓扑替康是拓扑异构酶Ⅰ抑制剂,可特异性与DNA单链断端上的拓扑异构酶Ⅰ相结合,阻止拓扑异构酶Ⅰ对单链断端的修复,破坏DNA双链结构,从而导致细胞死亡。Lee等采用去甲氧柔红霉素每天10mg/m 2 ,d1~3、Ara-C 1g/m 2 ,q12h,d1~5、拓扑替康1.25mg/m 2 ,d1~5,治疗难治/复发AML40例,CR率为59%,中位CR率和生存期分别为6个月和12个月。
以小剂量阿克拉霉素和阿糖胞苷联合G-CSF组成的CAG方案,20世纪90年代由日本学者设计报道,治疗难治和复发、继发AML,CR率分别达到87%和62%。其原理是AML细胞表达G-CSF和GM-CSF受体,G-CSF可预激(priming)处于G 0 期的白血病细胞进入增殖周期与化疗药物接触,从而增强抗白血病的疗效。由于本方案中阿克拉霉素和阿糖胞苷的剂量明显低于常规剂量,因此毒副作用相对较小。该方案不仅适用于难治和复发AML,也可适用于老年及低增生AML患者。有报告认为,CAG方案中加入地西他滨可进一步提高疗效。
(3)难治性ALL的治疗:
无论是难治或复发ALL对化疗药物均有不同程度的耐受性,对常规联合化疗反应皆不满意,预后较差,是当前ALL治疗中的最为棘手的问题之一。虽然50%的复发性ALL使用原诱导缓解方案仍有效,但再度缓解期极短。与AML相似,复发病例的疗效与上次缓解期的长短有关:第1次缓解期越长,获第2次缓解的概率越高,完全缓解后持续时间也越长。复发后病情严重患者很少能再次CR,即使缓解,极少(<5%)能长期存活。ALL患者的复发部位如在髓外,如中枢神经系统或睾丸等,预后更差。
难治/复发Ph + ALL首先应检测是否发生 ABL 基因突变。伊马替尼耐药患者,如有 Y253 H 、 E255 K / V 、 F359V / C / I 突变可选择达沙替尼(Dasatinib); F317L / V / I / C 、 T315A 、 V299 L 突变可选择尼洛替尼(Nilotinib);泊沙替尼(Bosutinib)可用于除 T315I 突变以外的对伊马替尼耐药的患者; T315I 突变可选择泊那替尼(Ponatinib)。Omacetaxinemepesuccinate(商品名Synribo)是 Teva Pharmaceuticals研制的半合成高三尖杉酯碱,临床试验结果显示对CML T315I 突变患者有效。2012年9月被美国食品药品管理局(FDA)加速批准用于慢性粒细胞白血病 T315I 突变患者的临床治疗,2014年2月已获得完全批准。诱导治疗期,每天皮下注射2次Synribo,每次1.25mg/m 2 ,连续14天,1个周期为28天。获得疗效后,维持治疗用法为1.25mg/m 2 ,每天2次,每个周期连续7天。体外实验表明,Synribo对Ph + ALL T315I 突变细胞也有效,尚有待临床试验结果证实。
难治/复发Ph - ALL AYA组患者如复发时间距首次诊断已超过3年,可试用初次诱导方案。其他Ph - ALL患者可选择含氟达拉滨或氯法拉滨的方案。增大剂量的hyper-CVAD方案也可使用。去甲氧柔红霉素联合大剂量Ara-C的CR率为44%。鬼臼类药物VM26或VP16、安丫啶与大剂量Ara-C亦有协同作用。FLAG方案对复发或难治性ALL均有效。奈拉滨(nelarabine)化学名为9β-D-阿拉伯呋喃糖-6-甲氧基-9H-嘌呤-2-胺。奈拉滨可在腺苷脱氨酶作用下,去甲基转化成ara-G,在脱氧鸟苷激酶和脱氧苷激酶作用下单磷酸化,转化为活性5-三磷酸盐ara-GTP。Ara-GTP在白血病细胞中蓄积到一定程度后嵌合入DNA中,从而抑制DNA的合成。由于ara-GTP在T细胞内比在B细胞内的累积速度更快,累积量更多,对T细胞有更强的选择性细胞毒作用。奈拉滨1.5g/(m 2 ·d),d1、d3、d5静脉输注,治疗难治性T-ALL患者,CR率可达31%,整体反应(OR)率41%。Binatumomab(商品名 Blincyto)是2014年新上市的单克隆抗体类药物,Binatumomab除了选择性地靶向作用于B细胞表面抗原CD19外,还可以特异性地结合T细胞表面抗原CD3,从而激活T细胞。临床试验结果表明Binatumomab对于难治性B-ALL有效。
异基因造血干细胞移植后复发的ALL患者可以考虑实施第二次异基因移植或供体淋巴细胞输注(DLI)。
(四)中枢神经系统白血病的预防与治疗
随着急性白血病缓解率提高和存活期延长,中枢神经系统白血病的发生率也明显增多。目前所用抗白血病药物在常规剂量下多数不能通过血脑屏障,故中枢神经系统成为白血病细胞的隐蔽所,常为急性白血病复发的重要根源,应加强防治。
ALL患者初诊时中枢神经系统累及的比例约为3%~7%。在治疗过程中如果不作中枢神经系统直接治疗(CNS-directed therapy),最终中枢神经系统白血病的发生率可>50%。因此,应常规实施中枢神经系统白血病的预防措施。标准方法是鞘内注射抗白血病药物。通常在诱导缓解一开始或CR后,立即在鞘内注射MTX,每次10mg,每周2~3次。大剂量Ara-C或大剂量MTX全身化疗能使药物透过血脑屏障,对中枢神经系统白血病也有肯定的预防作用。低危ALL的预防措施可采用大剂量全身化疗+4次鞘内化疗,高危ALL为大剂量全身化疗+8次鞘内化疗,成熟B-ALL或Burkitt白血病则须将鞘内注射增至16次。美国NCCN肿瘤临床实践指南推荐ALL中枢神经系统的评估状态分为3级:CNS-1:脑脊液无论白细胞计数多少,未发现幼稚淋巴细胞;CNS-2:脑脊液存在幼稚淋巴细胞,但白细胞<5/mm 3 ;CNS-3:脑脊液存在幼稚淋巴细胞,白细胞≥5/mm 3 。评估时为排除穿刺损伤因素,强调对于脑脊液检查结果评级虽为CNS-3,但外周血存在白血病细胞的患者应作外周血和脑脊液WBC/RBC比值的比较。脑脊液的比值至少是外周血的2倍以上,才可以确定为CNS-3,否则为CNS-2。
AML患者至今尚无统一的规定。一般认为,M 3 、M 4 、M 5 患者以及所有初诊时外周血存在白血病细胞的患者应在CR后常规行腰穿做脑脊液检查,并预防性鞘内注释化疗药物。难治复发ALL患者,无论Ph染色体是否阳性,均需要进行鞘内化疗。
确诊为中枢神经系统白血病(central nervous system leukemia),治疗方法有以下几种:
1.糖皮质激素
主要控制中枢神经系统白血病的症状。地塞米松10mg静脉注射2~3天,可使头痛、呕吐等症状减轻,但脑脊液、脑神经瘫痪及神经乳头水肿无明显改善。
2.甲氨蝶呤鞘内注射
以10~15mg,每周2~3次鞘内注射,直至脑脊液白血病细胞完全清除为止。本法能较快控制中枢神经系统白血病,但缓解期短,容易复发。所以中枢神经系统白血病缓解后应继续每周1次鞘内注射用甲氨蝶呤5~10mg,连续4~6周。鉴于甲氨蝶呤经鞘内注射,在脑室内浓度常不易达到抗肿瘤作用,现设计有皮下脑脊液贮存器(ommaya reservoir),将甲氨蝶呤直接注射至脑室。Bleyer等将脑室和鞘内甲氨蝶呤注射作了比较,前者治疗效果较好。但脑脊液贮存器安装后约18%病例有出血、阻塞和继发感染等并发症。脑脊液贮存器用于中枢神经系统白血病为髓外复发的病例较为合适。
甲氨蝶呤鞘内注射后可引起急性化学性蛛网膜炎和亚急性脑和脊髓运动神经元功能不良等毒性作用。患者可有头痛、发热或呕吐,出现于第1~10次注射期间。如不停药,反应可逐渐加重。曾报道有7例ALL中枢神经系统白血病在治程中或停药后不久发生痴呆、神经错乱、易激惹、嗜睡、共济失调、癫痫发作,其中有2例昏迷,1例死亡。另有报道在注射甲氨蝶呤后发生意外者共7例,表现有感觉障碍伴轻度运动功能减退,下肢或四肢瘫痪等,其中死亡者也有2例。意外反应常突然发生,或出现在鞘内注射0.5~24小时内。上述毒性反应可能与甲氨蝶呤的保存液羟基甲酸或稀释液甲醇有关,它们能阻断神经纤维传导,也可使神经纤维脱髓鞘。个别病例可能是机体对甲氨蝶呤产生急性变态反应。甲氨蝶呤可通过脑膜吸收而产生全身反应,应加注意。骨髓已受到抑制或肾功能不全更应慎用。鞘内注射药物容积一般为脑脊液的10%,即10~15ml。当脑脊液压力过高时,应酌情减量。注射应缓慢,有反应时随时停药。如有条件监测脑脊液内甲氨蝶呤浓度,可减少甲氨蝶呤神经毒性反应的发生率。
3.阿糖胞苷鞘内注射
甲氨蝶呤鞘内注射有抗药者,可用阿糖胞苷25mg/m 2 ,每周2~3次,鞘内注射;也可采用MTX、Ara-C与地塞米松联合鞘内注射。
4.头颅与脊髓照射
仅用颅脑 60 Co或直线加速器照射(5~10Gy)只能缓解症状,不能使脑脊液恢复正常,缓解率也低。如果加用脊髓照射10Gy,效果较好,但对骨髓抑制作用比较明显。对于头颅CT/MRI检查发现肿块的AML患者,一般采用放疗后鞘内给药。ALL患者确诊为中枢神经系统白血病[CNS-3和(或)脑神经累及],建议接受剂量为18Gy的放疗。
(五)造血干细胞移植
1.异基因造血干细胞移植
AML和ALL均为异基因造血干细胞移植(allogeneic hemopoietic stem cell transplantation)的适应证。首次完全缓解期的AML患者,应当根据疾病细胞遗传学的特征来决定缓解后的继续治疗措施。预后好的患者可采用足够强度的化疗作为巩固治疗,5年总生存率可达50%以上。也可考虑自体造血干细胞移植。风险更大的异基因造血干细胞移植一般不作为该组患者的首选,可作为复发早期或第二次缓解期的治疗策略。对预后中等的患者,如有HLA匹配的家庭成员供者进行移植,3年无病生存率可达65%,3年复发率为18%。预后差组如有HLA匹配的家庭成员供体,应当在完全缓解后尽快行造血干细胞移植。在经过选择的病例中,如果在第一次缓解期就接受非血缘关系的HLA相匹配供者或家庭成员供者移植,长期生存率仍可达到40%~50%。
成人ALL复发率高,异基因造血干细胞移植在成人ALL的治疗中占据重要地位。2008年报道的一项国际协作临床试验(MRC UKALL Ⅻ/ECOG E2993)分析1993年至2006年1913例成人ALL的资料表明,Ph - ALL患者采用异基因造血干细胞作为缓解后的治疗措施,其5年总存活为53%,明显高于自体移植和化疗患者的45%。2002年IBMTR报告接受移植的2820名ALL患者资料显示,在CR1期移植,年龄<20岁与年龄>20岁组3年无病生存率分别为61%±4%和48%±4%;在CR2以上缓解期移植,3年无病生存率在年龄<20岁与年龄>20岁组分别为47%±6%和30%±5%;无关供者的移植在CR1或以后的缓解期进行3年无病生存率分别为45%±3%和36%±8%;处于疾病进展期的患者无病生存率为10%~15%。法国的一项大型多中心临床试验(LALA87)的资料显示,257例随机抽样的ALL病例中,116例接受异基因造血干细胞移植,对照组114例接受化疗或自体造血干细胞移植(autologous hemopoietic stem cell transplantation),两组的5年生存率差异无统计学意义。但在高危病例,异基因造血干细胞移植组5年总生存率和5年无病生存率分别为44%和39%,明显高于对照组的20%和14%。另有一项关于Ph + ALL的研究结果显示,167例接受造血干细胞移植,其中49例为HLA相配的相关供体移植,23例为HLA相配的无关供体移植,7例为自体造血干细胞移植。77例接受持续化疗。5年的疾病复发危险性,异基因造血干细胞移植组为29%,明显低于自体造血干细胞移植/化疗组的81%。而5年生存率异基因造血干细胞移植组为43%,自体造血干细胞移植/化疗组为19%。因此,目前较为一致的观点是对于Ph + ALL患者,尽可能争取在首次缓解后实施异基因造血干细胞移植。
2.自体造血干细胞移植
(1)AML:
2002年来自希腊的120例临床病例研究显示,年龄≤60岁的AML患者,自体造血干细胞移植的疗效明显不如异基因造血干细胞移植,3年无失败生存率(FFS)分别为42%和73%,与大剂量Ara-C巩固治疗比较也不能显示其优势。以往认为对于具有良好细胞遗传学预后因素的AML患者,自体造血干细胞移植的疗效优于单纯化疗,但近年来随着抗白血病新药的出现和化疗方案的改进,尤其是大剂量阿糖胞苷等在巩固强化治疗阶段中的应用,自体造血干细胞移植在该组AML中的地位受到质疑,目前国外一些临床研究中心有放弃将自体造血干细胞移植作为首次缓解后的一线治疗措施的趋势。对于具有中等细胞遗传学预后因素的AML患者,由于复发率较预后良好组患者显著为高,如无异基因造血干细胞移植的合适供体,可考虑行自体造血干细胞移植。国外的一项资料显示,该组患者5年生存率自体造血干细胞移植为56%,单纯化疗为48%。具有不良细胞遗传学预后因素的AML患者,自体造血干细胞移植疗效欠佳,5年生存率仅为15%,远低于异基因造血干细胞移植的疗效。对于60岁以上的老年AML患者,最近来自EORTC-Gimema AML-13临床试验的资料表明自体外周血干细胞移植(autologous peripheral stem cell transplantation)亦不能改善其预后。不过也有持不同观点的研究结果。
(2)ALL:
国外多项临床资料表明,成人ALL自体造血干细胞移植的疗效明显较异基因造血干细胞移植为差。法国的大型多中心临床试验(LALA87)数据表明,无论高危和标危ALL患者,自体造血干细胞移植与化疗比较都不能显示其优势。Anderson癌症中心的资料也持类似的观点。2008年报道的MRC UKALLⅫ/ECOG E2993临床试验甚至得出自体造血干细胞移植不如化疗的结论。欧洲骨髓移植组曾报道510例ALL患者行自体骨髓移植的疗效,CR1期和CR2期的7年无病生存分别为50%和20%,其中CR1期在诊断40天内达CR者其无病生存较40天以上达CR者显著增高,分别为60%和30%。从这项结果可以看出自体造血干细胞移植治疗ALL的时机应选择CR1期,其疗效与白血病细胞对化疗药物的敏感性相关。
主要参考文献
1.林果为,欧阳仁荣,陈珊珊,等.现代临床血液病学.上海:复旦大学出版社,2013:838-886.
2.Lee GR,Foerster J,Luken J,et al.Wintrobe's Clinical Hematology.13th ed.Baltimore:Williams&Wilkins,2014:1548-1672.
3.Hoffman R,Benz Ir EJ,Siberstein LE,et al.Hematology:Basic principles and Practice.6th ed.Philadephia:Elsevier saunders,2013:853-881,960-980.
4.NCCN Guidelines.Acute myeloid leukemia.Version 2.2016.
5.NCCN Guidelines.Acute lymphoblastic leukemia.Version 1.2016.
6.Goldstone A H,Richards SM,Lazarus HM,et al.In adults with standard-risk acute lymphoblastic leukemia,the greatest benefit is achieved from a matched sibling allogeneic transplantation in first complete remission,and an autologous transplantation is less effective than conventional consolidation/maintenance chemotherapy in all patients:final results of the International ALL Trail(MRC UKALLⅫ/ECOG E2993).Blood,2008,111(4):1827-1833.
7.Willemze R,Suciu S,Meloni G,et al.High-dose cytarabine in induction treatment improves the outcome of adult patients younger than age 46 years with acute myeloid leukemia:results of the EORTC-GIMEMA AML-12 trial.J Clin Oncol,2014,32(3):219-228.
8.Burnett AK,Russell NH,HillsRK.A randomized comparison of daunorubicin 90mg/m 2 vs 60mg/m 2 in AML induction:results from the UK NCRI AML17 trial in 1206 patients.Blood,2015,125(25):3878-3885.
9.Curran E,Stock W.How I treat acute lymphoblastic leukemia in older adolescents and young adults.Blood,2015,125(24):3702-3710.
10.Rytting ME,Jabbour EJ,Jorgensen JL,et al.Final results of a single institution experience with a pediatric-based regimen,the augmented Berlin-Frankfurt-Münster,in adolescents and young adults with acute lymphoblastic leukemia,and comparison to the hyper-CVAD regimen.Am J Hematol,2016,91(8):819-823.
第三节
慢性白血病
陈 彤
慢性白血病(chronic leukemia)是一组异质性造血系统肿瘤,病程较缓慢,白血病细胞有一定的分化成熟能力,骨髓及周围血中以异常的较成熟细胞为主。临床上有两种类型:①慢性粒细胞白血病(chronic myelogenous leukemia,chronic myelocytic leukemia or chronic granulocytic leukemia,CML);②慢性淋巴细胞增殖性疾病(chronic lymphoproliferative disorders,CLPD),包括慢性淋巴细胞白血病、幼淋巴细胞白血病、毛细胞白血病、绒毛淋巴细胞脾淋巴瘤、大颗粒淋巴细胞白血病、成人T细胞白血病/淋巴瘤、Sézary综合征等。CLPD再根据免疫表型分成B细胞型、T细胞和NK细胞型。
慢性粒-单核细胞白血病、不典型慢性粒细胞白血病、幼年型粒-单核细胞白血病、慢性中性粒细胞白血病、慢性嗜酸性粒细胞白血病也均属于慢性白血病。WHO分型已将其分别归入骨髓增生异常/骨髓增殖性肿瘤,可参见本篇第三章和第四章。
一、慢性粒细胞白血病
慢性粒细胞性白血病简称慢粒,是起源于多能造血干细胞的恶性克隆增殖性疾病,表现为髓系各个阶段细胞的过度增殖,以外周血中性粒细胞增多并出现幼稚粒细胞、嗜碱性粒细胞增多、贫血、血小板增多和脾大为特征,具有异常的Ph染色体t(9;22)(q34;q11.2)和 BCR-ABL1 融合基因,可从慢性期(chronic phase,CP)向加 速期(accelerated phase,AP)、急变期(blastic phase,BP或 blast crisis,BC)发展,一旦转变为急性白血病,预后较差。
慢粒约占全部白血病的15%,国内慢性白血病中90%为慢粒,发病年龄大多在20~60岁,发病率随年龄的增长逐步上升,45~50岁年龄组最高,5~20岁仅占慢粒的10%以下,男性略多于女性。我国慢粒的年发病率约为0.36/10万,患者确诊时中位年龄40.02(2.45~83.29)岁,男女比例约为1.78∶1。
【病因与发病机制】
大剂量的放射线照射是慢粒较明确的致病因素。日本广岛和长崎原子弹爆炸后幸存者、英国强直性脊柱炎患者接受放疗后以及宫颈癌放疗的患者中,慢粒的发病率明显高于普通人群。
慢粒是一种获得性、起源于单个干细胞的肿瘤性疾病。90%以上的慢粒患者中可发现有Ph染色体,即t(9;22)(q34;q11),9号染色体q34带上原癌基因 c-abl 的片段易位至22号染色体q11带上的断裂点簇集区bcr(break point cluster region),产生 BCR-ABL 融合基因,转录成融合mRNA,编码生成具有很强酪氨酸蛋白激酶活性的融合蛋白,参与细胞信号传导途径中的多种蛋白磷酸化,抑制细胞凋亡,削弱造血祖细胞与骨髓基质细胞的黏附,使细胞生长缺乏接触抑制而致增殖过度(图16-3-1)。
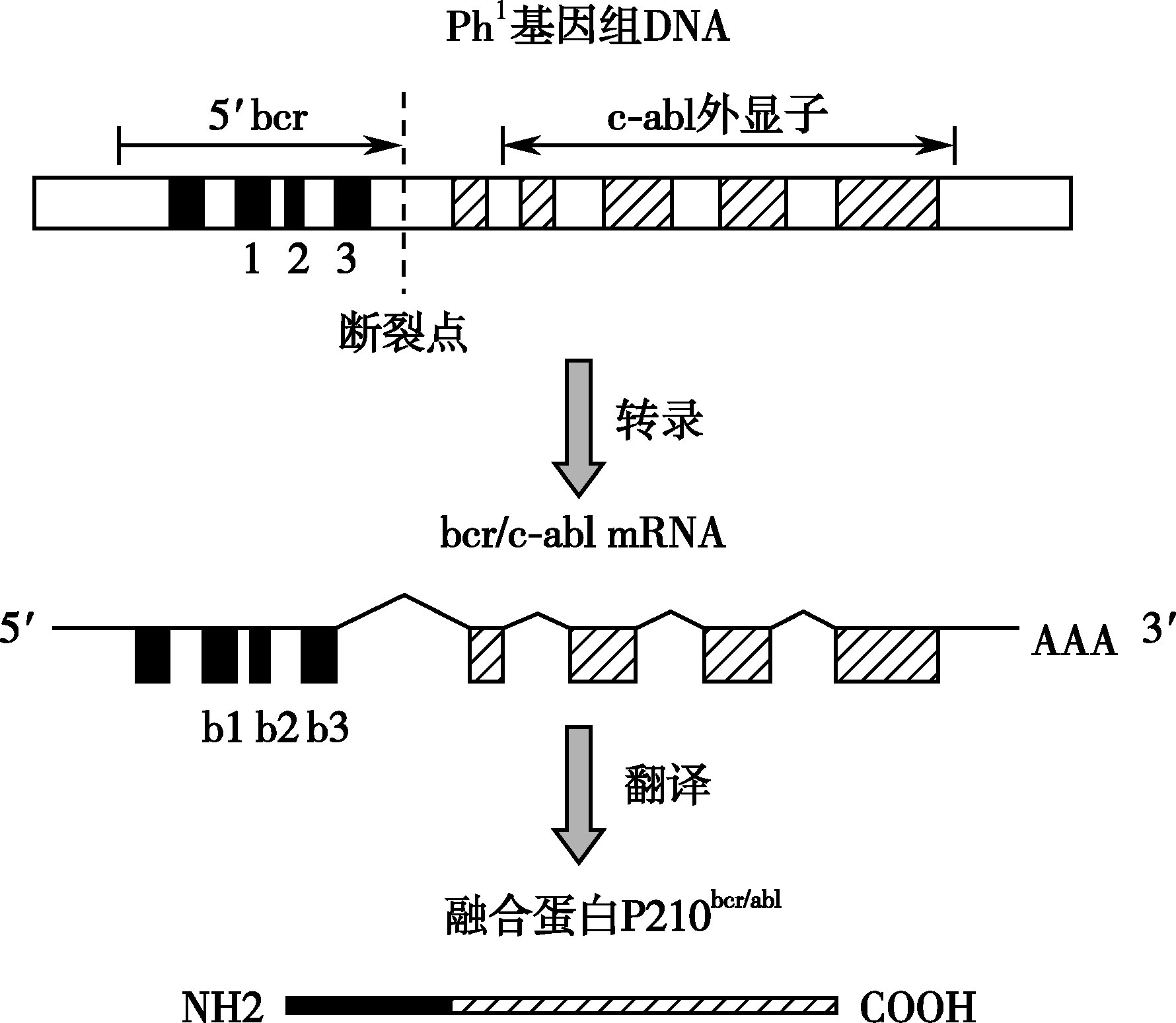
图16-3-1 BCR - ABL 融合基因示意
22号染色体上的BCR位点主要有三种:M-bcr、m-bcr和μ-bcr,分别形成3种融合蛋白P210、P190和P230。大部分慢粒患者在e14a2或e13a2位点融合,表达P210融合蛋白。而P190(e1a2)和P230(e19a2)分别主要与Ph染色体阳性的急性淋巴细胞白血病和慢性中性粒细胞白血病的发生相关。
【临床表现】
起病缓慢,症状多为非特异性,绝大多数患者起病时处于慢性期。患者可因造血过盛的症状和体征就诊,如易疲倦、乏力、食欲缺乏、低热、多汗、体重减轻、上腹部不适及脾大。大约10%~30%的患者在出现症状前因定期体检而发现,起病时即处于加速期或急变期的患者各占10%左右。
1.脾大
脾大程度不一,与外周血白细胞升高的水平有关,大约50%以上的患者确诊时脾可大至肋缘下10cm以上,质坚无压痛,患者常感上腹部饱胀不适。少数患者因发生脾梗死或脾周围炎而出现显著左上腹和左肩部疼痛,可有局部压痛和摩擦音,自发性脾破裂罕见。15%~20%的患者有肝大,程度较轻。淋巴结肿大较少见,但可作为早期急变的首发症状。
2.发热、贫血和出血
由于肿瘤负荷增加,可出现典型的怕热、消瘦和盗汗等高代谢综合征。疾病早期甚少有感染,白细胞黏附、游走、吞噬等功能下降的缺陷可由于细胞数量增加而得到补偿。血小板聚集功能下降,但明显的贫血及出血多在急变期才出现。
3.白细胞淤滞综合征
较少见。当白细胞极度增高时,由于白细胞淤滞、循环受阻,可出现呼吸困难、发绀、脏器梗死、眼底静脉扩张、视乳头水肿、眼底出血和阴茎异常勃起、耳鸣、神志改变,甚至中枢神经系统出血等表现。
4.其他
胸骨压痛较常见,多在胸骨下段。细胞破坏、血尿酸升高引起痛风性关节炎。嗜碱性粒细胞增多,组胺释放出现荨麻疹、皮肤瘙痒以及消化性溃疡。皮肤浸润较少见,偶有中性粒细胞浸润至真皮层而表现为急性发热性中性粒细胞皮病(Sweet综合征)。
【实验室检查】
1.血常规
外周血中白细胞升高是主要的特征,通常>25×10 9 /L,半数患者在100×10 9 /L以上。分类可见各期粒细胞,中性晚幼及杆状核粒细胞的比例明显增多,原粒和早幼粒细胞较少,可见过度分叶核粒细胞。嗜酸及嗜碱性粒细胞绝对值均可增多,嗜碱性粒细胞的比例可以指导慢粒的分期诊断,慢性期多在10%~15%以下。确诊时红细胞数大多正常或轻度变化,少数可出现红细胞形态异常,并可见到少量有核红细胞,网织红细胞计数正常或轻度增多。大约50%的患者确诊时血小板计数高于正常,在慢性期可逐渐升高。若血小板计数明显升高或降低,则预示着疾病向加速期或急变期进展。
2.骨髓象
有核细胞增生极度活跃,以粒系增生为主,造血组织占整个骨髓体积的75%~90%,脂肪含量明显减少。红系增生受抑,粒/红比值可达10~30∶1,原粒和早幼粒细胞一般不超过5%~10%,嗜酸及嗜碱性粒细胞比例增多。巨核细胞数量正常或增加,半数患者骨髓内Ⅲ型胶原(网状纤维)增生,部分可发生骨髓纤维化。
3.祖细胞集落培养
慢性期骨髓和外周血粒系、巨核系、嗜酸粒系集落形成增加,分别为正常的20倍和500倍左右。具有长期造血能力的原始祖细胞亦显著增加,所形成的集落较正常致密。进入加速期和急变期后祖细胞的增殖和分化能力减弱,集簇增加,已成为慢粒的分期指标之一。
4.中性粒细胞碱性磷酸酶测定
90%以上的患者成熟中性粒细胞碱性磷酸酶(NAP)积分降低或缺失,治疗后白细胞下降或接近正常,炎症感染时该酶活性可升高或接近正常。NAP检测有助于与类白血病反应及其他骨髓增殖性疾病相区别,也可作为预后指标。
5.细胞遗传学检测
90%以上的慢粒患者可发现Ph染色体t(9;22)(q34;q11),是慢粒的标记染色体。Ph染色体存在于有核红细胞、粒细胞、单核细胞、巨核细胞以及T、B淋巴祖细胞中,但并不见于外周血T、B淋巴细胞中。在慢粒慢性期,大约70%的患者为典型的t(9;22)(q34;q11),另有20%的患者可表现为特殊的核型,如[t(Ph),22q - ]、[t(Ph),-Y]、[t(Ph),+8]等。当进入加速期或急变期时,约75%的患者合并Ph染色体以外的染色体核型异常,大约5%患者可出现累及三条染色体的复杂易位。
6.分子生物学检测
通过FISH、RT-PCR、Southern blotting、Western blotting等技术对t(9;22)分子序列的检测可以提供基因重排的依据,补充细胞遗传学在诊断上的不足,对Ph染色体阴性的慢粒有进一步确诊价值。FISH利用5'-BCR和3'-ABL1探针可以检测 BCR-ABL 融合,假阳性率在1%~10%。定量PCR技术可从10 5 ~10 6 正常细胞中检测出一个融合基因阳性的肿瘤细胞,对于治疗后Ph染色体转阴患者进行微量残留病灶的检测有很大价值,也可用于明确患者有无分子水平复发。实时定量PCR是国际上检测 BCR-ABL 转录本最常用的方法,但需注意假阳性的发生。
7.血清生化测定
由于粒细胞中有维生素B 12 结合蛋白,慢粒时血清维生素B 12 和维生素B 12 结合力均显著增高,维生素B 12 值可达正常的10倍以上,且与白细胞值呈正相关,缓解期血清维生素B 12 浓度可下降但仍高于正常。血清尿酸、乳酸脱氢酶浓度也均增高,化疗后因粒细胞破坏而更为明显。
【诊断与鉴别诊断】
根据典型的外周血白细胞增高以及分类异常、嗜碱性粒细胞绝对计数增高、脾大伴有Ph染色体或其变异核型以及22号染色体上的 BCR-ABL 基因重排,诊断并不困难。
本病应与以下疾病鉴别:
1.反应性白细胞增多(类白血病反应)
多发生在严重感染、肿瘤或炎症性疾病的基础上,无Ph染色体和 BCR-ABL 融合基因,外周血白细胞可达(30~100)×10 9 /L,以中性杆状核居多,可有少量晚幼粒细胞,原始及早幼粒细胞罕见,中性粒细胞NAP积分升高或正常。
2.其他慢性骨髓增殖性肿瘤(cMPN)
慢粒可合并骨髓纤维化,也可同时有血小板和红细胞增多,因此需与其他骨髓增殖性疾病,如真性红细胞增多症(真性红细胞增多症)、原发性血小板增多症(ET)、原发性骨髓纤维化(MF)等鉴别。一般来说,90%的慢粒患者白细胞计数持续在30×10 9 /L以上,而其他cMPN常以某一系细胞异常增多为特征,白细胞一般在30×10 9 /L以下,无Ph染色体和 BCR / ABL 融合基因,且有相应病变的表现。真性红细胞增多症表现为红细胞的显著增高;ET血小板计数在450×10 9 /L以上,中性粒细胞仅轻中度增高(<20×10 9 /L)。MF以外周血中出现泪滴样红细胞和有核红细胞为特征。近来,cMPN在分子生物学诊断方面有很大的进展。95%的真性红细胞增多症患者和40%~50%的MF、ET患者有 JAK2 基因突变,有助于和慢粒的鉴别。
3.其他类型慢性髓系白血病
随着对其他慢性髓系白血病的深入了解,原来对Ph阴性慢粒的诊断需要进一步修正。①慢性粒单核细胞白血病(CMML):CMML属于MDS/MPN范畴,外周血单核细胞持续性增高>1×10 9 /L,中性粒细胞碱性磷酸酶积分正常或增高,无Ph染色体和 BCR-ABL 融合基因。②不典型慢性粒细胞白血病(aCML):临床表现类似Ph染色体阳性CML,但嗜碱性粒细胞无明显增多,骨髓血细胞可具有病态造血的形态学表现,无Ph染色体和 BCR-ABL 融合基因,对治疗CML的药物反应较差,病程进展快。
4.其他
慢粒有贫血及脾大时需与肝硬化、血吸虫病、淋巴瘤等鉴别,发生脾梗死及脾周围炎时应与急腹症相鉴别。
【临床分期】
慢粒可分为慢性期(CP)、加速期(AP)和急变期(BP)。各期的诊断标准如下:
1.慢性期
①无症状或有低热、乏力、多汗、体重减轻等症状;②白细胞计数增高,主要为中性中、晚幼和杆状核粒细胞。原始粒细胞(Ⅰ型+Ⅱ型)<5%~10%,嗜酸性粒细胞和嗜碱性粒细胞增多,可有少量有核红细胞;③骨髓增生明显至极度活跃,以粒系增生为主,中、晚幼粒细胞和杆状核粒细胞增多,原始粒细胞<10%;④有Ph染色体或 BCR-ABL 融合基因;⑤CFU-GM培养集落和集簇较正常明显增加。
2.加速期
具有下列之一者可诊断为本期:①持续性的外周血白细胞增高>10×10 9 /L或进行性脾大,治疗无效;②对治疗无反应的血小板持续增高(>1000×10 9 /L);③与治疗无关的血小板进行性降低(<100×10 9 /L);④出现克隆演变的遗传学证据(即慢粒初诊时没有的其他遗传学异常);⑤外周血嗜碱性粒细胞>20%;⑥外周血或骨髓中原始细胞占10%~19%。标准①~④常提示疾病从CP向AP的转变,标准⑤和⑥更多见于AP向BP的发展。
3.急变期
使用伊马替尼治疗慢粒后可以显著延缓疾病进展,延长患者的慢性期,但CML干细胞在酪氨酸激酶作用下并不产生凋亡,疾病因克隆演变向急性白血病转变的危险仍旧存在。具有下列之一者可诊断为本期:①外周血或骨髓中原始细胞>20%;②髓外原始细胞增殖。慢粒急变通常为急粒变或急粒单变,约10%的患者可出现红白血病变,偶见巨核细胞白血病变、早幼粒变或嗜碱粒变,1/3的患者可急淋变,有些病例可呈粒淋双表型变。一旦急变后,往往在3~6个月内死于各种并发症。
【治疗】
对所有的CML初诊患者进行细胞遗传学或分子学检测,评估CML诊断时疾病分期对CML治疗选择非常重要。而对治疗选择的分层及基于规范监测的治疗方式的适时转化,对于改善慢粒患者的生存尤为重要。
(一)慢性期治疗
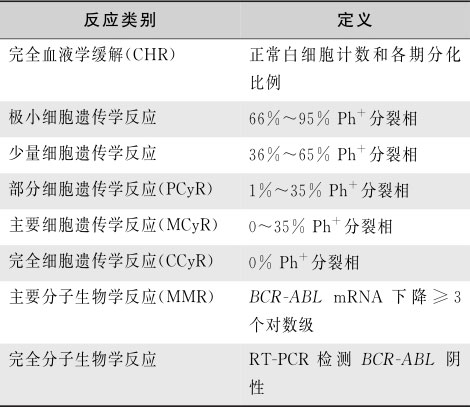
治疗目的是促进正常造血干/祖细胞的生长和抑制白血病克隆增殖,以期降低外周血白细胞计数,缓解脾大并控制高代谢综合征,达到分子生物学完全缓解。治疗后血液学完全缓解的标准包括外周血细胞计数正常,白细胞计数<10×10 9 /L、血小板计数<450×10 9 /L、外周血无幼稚细胞、无脾大的症状和体征。细胞遗传学缓解标准根据骨髓中细胞分裂中期Ph染色体的比例决定。分子生物学缓解标准根据骨髓或外周血中 BCRABL1 转录本下降的对数级来决定(表16-3-15)。
表16-3-15 慢粒治疗反应的标准和定义
1.药物治疗
(1)酪氨酸激酶抑制剂(TKI):
酪氨酸激酶抑制剂可作为三磷酸腺苷(ATP)与酪氨酸激酶结合的竞争性抑制剂,也可作为酪氨酸的类似物,阻断酪氨酸激酶的活性,抑制细胞增殖,进而达到治疗慢粒的目的。目前临床上最常用的针对BCR-ABL酪氨酸激酶小分子抑制剂(TKI)有甲磺酸伊马替尼、达沙替尼和尼罗替尼。
甲磺酸伊马替尼(格列卫,Glivec/Gleevec,STI571)为2-苯胺嘧啶衍生物,是ABLl特异性酪氨酸激酶的抑制剂,能特异性阻断ATP在ABL激酶上的结合位置,使酪氨酸残基不能磷酸化,从而抑制 BCR-ABL 阳性细胞的增殖(图16-3-2)。口服后生物利用度达98%,半衰期18小时,属于慢粒诱导缓解类药物,是治疗慢粒的首选药物。
伊马替尼具有较高的血液学完全缓解(CHR)和细胞遗传学完全缓解(CCyR)率(表16-3-15)。慢性期口服用量400mg/d,如果以常规剂量未能获得细胞遗传学和分子生物学缓解,或者疾病处于进展阶段可增至600~800mg/d。以400mg/d治疗的患者中,>75%的患者可以获得主要细胞遗传学反应,50%以上的患者用药6个月内可以获得主要分子生物学反应,5年总体生存率和无疾病进展生存率分别达90%和93%。
伊马替尼治疗后可出现恶心、呕吐、水肿、肌肉痉挛、皮疹、骨痛等副作用,可适当应用镇吐、利尿剂或调整剂量。大约30%的慢性期患者使用伊马替尼后可出现3~4级的骨髓抑制,在加速期或急变期的患者中更为多见。对于慢性期患者,若中性粒细胞<1×10 9 /L或血小板低于50×10 9 /L,建议短暂停用伊马替尼,待中性粒细胞达到1.5×10 9 /L、血小板计数达到100×10 9 /L时再恢复伊马替尼治疗。这类患者可以400mg/d的剂量继续治疗,如果前次骨髓抑制的恢复时间>7天,恢复起始剂量推荐为300mg/d,可逐步调整至400mg/d,但不推荐以低于300mg/d的剂量维持。大约3%的患者在接受治疗的6个月内可出现肝脏损害,偶有脾破裂、脑水肿、视网膜水肿导致的视物模糊、严重水钠潴留、免疫性溶血性贫血、骨代谢异常等不良反应的报道。约15%的患者可出现皮肤过敏等反应,除了重症者(如剥脱性皮炎、多型红斑等)需要永久停药外,轻、中度皮疹患者仅需联用肾上腺糖皮质激素或短暂停药即可控制。
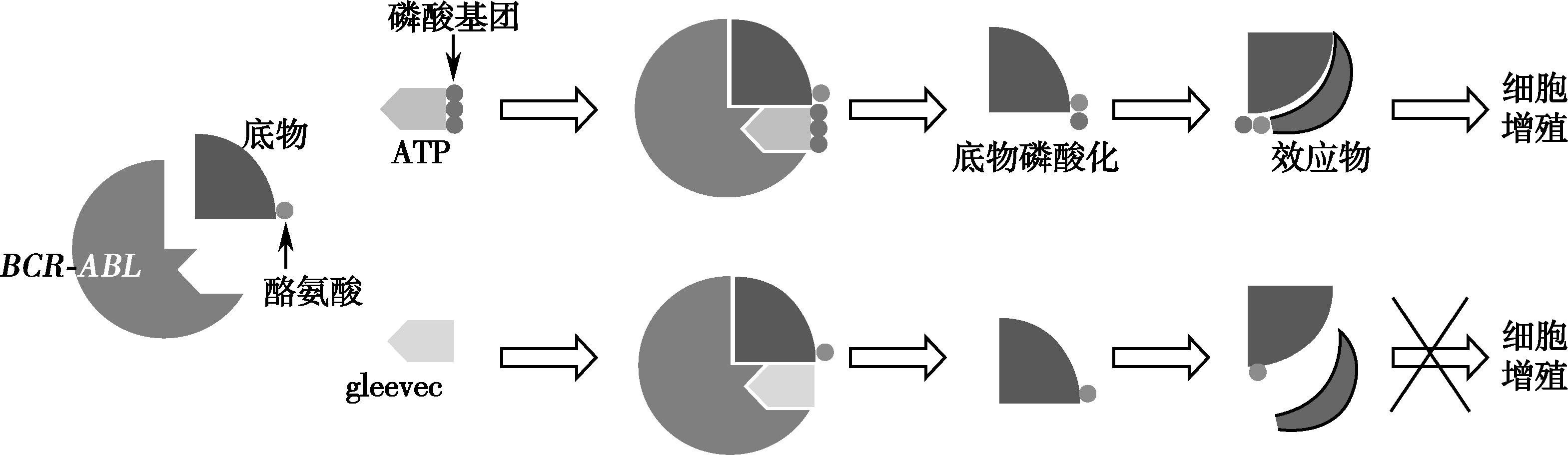
图16-3-2 甲磺酸伊马替尼作用机制示意
甲磺酸伊马替尼作为一线药物在治疗CML上取得了巨大成功,但仍有15%~25%的慢性期CML患者对伊马替尼耐药或不耐受,可使用第二代酪氨酸激酶抑制剂达沙替尼(dasatinib)或尼洛替尼(nilotinib)。目前 NCCN指南中推荐将达沙替尼(慢性期100mg qd、进展期140mg qd)和尼洛替尼(慢性期300mg bid、进展期400mg bid)用于对伊马替尼耐药或不耐受的CML患者的一线和二线治疗。
慢粒治疗反应的评判标准和TKI治疗后随访标准见表16-3-15。根据NCCN指南,在TKI起始治疗后每3个月随访1次,满半年后每6个月随访1次。使用伊马替尼最初半年内 BCR-ABL1 ≤10%或≥PCyR,继续同剂量TKI治疗;若 BCR-ABL1 ≥10%或未达到PCyR,3个月内的患者可考虑换用其他TKI或增加伊马替尼剂量至800mg/d,治疗满6个月的患者则需考虑换用其他TKI。TKI治疗1年以上的患者随访标准未满足CCyR者,均需考虑增加剂量或者换用其他TKI。
(2)干扰素-α(interferon-α):
干扰素-α可以直接抑制DNA多聚酶活性,治疗有效率与 BCR-ABL 的转录本数量有关。起始剂量可以为100万~300万U/d,隔日皮下注射,以后增加至500万U/d,每周3次,若能耐受,可增量至500万U/m 2 ,每日皮下或肌内注射1次,根据白细胞和血小板数量调节用量。使用干扰素-α早期有头痛、肌肉酸痛等流感样症状,延迟反应包括重要脏器功能受损、免疫性贫血、脱发、失眠、血小板减少和神经毒性等,约20%的患者对干扰素-α治疗不耐受。
(3)羟基脲(Hu):
是细胞周期特异性DNA合成抑制剂,毒性低,可延缓疾病进程,在TKI前是慢粒慢性期治疗的主要药物。开始剂量为1~3g/d,当白细胞降至20×10 9 /L时应减量至1~2g/d,此后随白细胞数量的变化调整剂量,维持量0.5~1.0g/d。单用本药不能清除Ph阳性细胞,并可使红细胞出现巨幼样改变。
(4)阿那格雷(anagrelide):
对血小板明显增高的慢粒患者可以使用阿那格雷,它可以减少巨核细胞数量降低血小板数量。对于以甲磺酸伊马替尼治疗后血小板仍持续在高水平的患者可以联用阿那格雷。
(5)其他药物:
其他包括白消安(Bu)、高三尖杉酯碱、靛玉红、甲异靛、巯嘌呤(6-巯基嘌呤)、6-硫鸟嘌呤、苯丁酸氮芥、环磷酰胺等,都可在一定程度上缓解慢粒的临床症状。
2.造血干细胞移植
自伊马替尼成功应用在慢粒的治疗后,采用造血干细胞移植手段治疗慢粒的例数已明显减少。对TKI治疗达到完全细胞遗传学缓解的初治慢性期患者一般不再主张进行异基因造血干细胞移植。而对于TKI治疗后复发、耐药、疾病进展至加速期或急变的患者,可考虑进行同种异基因骨髓或外周造血干细胞移植(allo-HSCT)。尤其在中国,由于单倍型造血干细胞移植体系的建立和完善、以及TKI在进展期患者中的应用,CML急变期行单倍型造血干细胞移植仍能获得良好疗效。在移植前是否应用TKI并不增加移植相关死亡率,但TKI疗效不理想常预示疾病进展。加速期或急变期患者进行allo-HSCT后使用伊马替尼TKI仍可获得细胞遗传学或分子生物学缓解。
3.白细胞单采
适用于白细胞数过高伴有白细胞淤滞综合征或妊娠患者,可缓解症状,降低化疗杀伤的白血病细胞数从而减少尿酸生成,但持续时间短、费用高。
4.放射治疗
脾区照射可用于化疗耐药、脾极度增大的患者,若有骨骼、软组织浸润也可采用局部放疗。
5.脾切除
适用于症状显著的巨脾或有脾功能亢进者,以提高输注血小板的疗效。但术后可能并发感染、栓塞或出血,甚至死亡。
(二)加速期和急变期治疗
对于加速期和急变期患者采用TKI单药或联合化疗,之后接受allo-HSCT已成为国内外推荐的标准治疗。未曾使用伊马替尼的患者可以选用伊马替尼桥接allo-HSCT治疗,而伊马替尼治疗过程中出现的疾病进展可以考虑达沙替尼、尼洛替尼和allo-HSCT。化疗方案根据细胞类型而定,急非淋变时可选用急性非淋巴细胞白血病的联合化疗方案,如中剂量Ara-C加米托蒽醌、去甲氧柔红霉素或依托泊苷(VP16)治疗;急淋变时按照急性淋巴细胞白血病的治疗方案。
(三)防止高尿酸血症的辅助治疗
慢粒确诊和复发时常伴有高尿酸血症,患者可出现痛风或肾脏损害,常随着治疗的开展而恶化。别嘌醇300mg/d,注意补充水分、利尿和碱化尿液等措施可以降低血尿酸。别嘌醇容易出现皮肤过敏现象,一旦出现应立即停药。血尿酸水平达9mg/dl以上时可考虑使用拉布立酶,疗效比别嘌醇显著。
【病程与预后】
在TKI广泛使用前,慢粒的中位数生存期为39~47个月,5年存活率为25%~35%。TKI的应用极大地延长了慢粒患者的无病生存时间。发病时外周血白细胞和血小板计数、原幼细胞比例、肝脾大小和嗜酸及嗜碱性粒细胞计数和慢性期长短与预后相关,通过这些预后指标预示治疗的反应性和生存时间。慢性期患者在TKI治疗过程中需通过染色体检查、FISH和定量PCR进行微小残留病灶的监测。70%的初发慢粒应用伊马替尼治疗12个月后可获得细胞遗传学缓解,使用60个月后这一比例可增至90%,对伊马替尼耐药或不耐受者可换用其他TKI。
二、慢性淋巴细胞白血病
慢性淋巴细胞白血病(chronic lymphocytic leukemia,CLL)简称慢淋,是一种慢性淋巴细胞增殖性疾病,以CD5 + 单克隆性B淋巴细胞在外周血、骨髓、脾和淋巴结等淋巴组织中大量克隆性积蓄为特征,细胞形态接近成熟淋巴细胞。慢淋的肿瘤细胞来源于记忆性B淋巴细胞,表面标志多为 CD19 + CD5 + CD23 + ,sIg、CD20、CD79b、FMC7的表达相对较弱。
我国慢淋发病率低,为0.05/10万,而在西方国家慢淋是最常见的成人白血病,构成比占所有白血病的20%~30%左右。男性发病率约为女性的2倍,大部分患者发病时年龄在50岁以上,中位年龄为65岁,30岁以下罕见。
【病因与发病机制】
环境和职业因素在B细胞慢淋的发病中并不占主要地位,长期接触低频电磁场可能和慢淋的发病有关。淋巴增殖性疾病家族史是慢淋的高危因素,发生率约占慢淋患者的1/10。大部分慢淋细胞处于非增殖期,细胞表达多种抗凋亡蛋白,具有较高的抗凋亡能力,细胞寿命较长而在外周血内聚积。
从细胞发生的角度可以将散发型CLL分为两种,一种高表达免疫球蛋白重链基因( IGHV )的突变,另一种则无 IGHV 突变。这两种慢淋的基因表达谱不同,其中ZAP-70的表达差异有助于两者鉴别。ZAP-70是一分子量为70×10 3 的Zeta相关蛋白,正常情况下表达在NK细胞或T细胞胞质内、与T细胞受体的 ζ链结合而具有蛋白激酶的活性。不具有 IGHV 基因突变的CLL细胞ZAP-70的表达较高,而具有 IGHV 基因突变的CLL细胞ZAP-70水平较低。 IGHV 基因突变和ZAP-70表达不同的慢淋,其细胞来源可能不同,来源于生发中心的慢淋,肿瘤细胞具有 IGHV 基因突变,ZAP-70表达低;来源于生发中心前的慢淋细胞则无 IGHV 基因突变,ZAP-70表达高。
【临床表现】
慢淋早期常无症状,患者常因发现无痛性淋巴结肿大或不明原因的淋巴细胞绝对值升高而就诊。患者有轻度乏力、易疲劳等非特异性表现,一旦进入进展期,除全身淋巴结和脾大外可表现为体重减轻、反复感染、出血和贫血症状。
1.淋巴结肿大
80%的患者确诊时有无痛性淋巴结肿大,可为全身性,轻至中度,偶见巨块型肿大,常累及颈部、锁骨上、腋下及腹股沟等处,口咽、泌尿道、胆道等部位的淋巴结过度肿大时可导致局部压迫。扁桃体、泪腺、唾液腺累及时,可产生Mikulicz综合征。
2.肝脾大
半数患者有轻至中度脾大,伴腹部饱胀感,晚期可达盆腔,可发生脾梗死或脾破裂。肿瘤细胞浸润引起的肝大少见。
3.贫血和出血
病情进展时可导致贫血或血小板减少而产生相应的症状,多数情况下由于白血病细胞骨髓浸润或产生自身抗体所致,偶见因脾大引起的脾功能亢进。溶血性贫血多见于温抗体型,抗体多为多克隆性,说明自身抗体并非完全由肿瘤细胞分泌;少数患者可出现纯红细胞再生障碍性贫血。
4.结外浸润
淋巴细胞可浸润至皮肤、结膜、肺、胸膜、胃肠道、骨骼、神经系统、肾脏、前列腺、性腺和眶后组织,但由浸润所致的症状并不多见。
5.并发症
由于低免疫球蛋白血症、补体水平低、T细胞功能缺陷以及免疫抑制剂的使用,患者的体液免疫和细胞免疫均受影响,而且慢淋白血病细胞可合成TGF-β等免疫抑制因子,因此大部分患者可合并免疫缺陷及免疫紊乱表现,如条件致病性病原体感染、自身免疫性疾病和第二肿瘤。
【实验室检查】
1.血常规
白细胞持续增多≥10×10 9 /L,淋巴细胞比例≥50%,单克隆淋巴细胞绝对值≥5×10 9 /L,部分患者确诊时白细胞可达100×10 9 /L。细胞形态接近正常的静止期淋巴细胞,胞质少、Wright-Giemsa染色呈蓝色,细胞核形态正常,偶见少数带核仁的幼稚淋巴细胞或不典型细胞。肿瘤细胞骨髓浸润、治疗后骨髓抑制、免疫破坏或营养元素缺乏等情况下可出现贫血或血小板减少。有20%的患者Coombs试验阳性,但仅有8%的患者出现自身免疫性溶血性贫血。部分患者可伴免疫性血小板减少性紫癜。
2.骨髓象
骨髓增生活跃,淋巴细胞显著增多,占40%以上,形态与外周血基本一致,原始淋巴细胞少见,红、粒及巨核细胞系生成受抑,有时呈纯红细胞再生不良。FAB依据形态将CLL分为三型:典型CLL(90%为小淋巴细胞);混合型(CLL/PL:幼淋巴细胞11%~54%);不典型CLL。典型CLL占80%。骨髓活检可判断骨髓受累的程度,分为间质型(30%)、结节型(10%)、结节-间质混合型(25%)和弥漫型(25%),后者提示病情进展迅速,预后较差。
3.淋巴结活检
淋巴结病理可见典型的小淋巴细胞弥漫性浸润,细胞形态与血液中的淋巴细胞一致,病理与低度恶性“小淋巴细胞淋巴瘤”的淋巴结病理表现类似,现WHO分型将慢性淋巴细胞白血病和小淋巴细胞淋巴瘤归成一类,称之为慢性淋巴细胞白血病/小淋巴细胞淋巴瘤(chronic lymphocytic leukaemia/small lymphocytic lymphoma,CLL/SLL)。少数患者可有少量散在分布的R-S样细胞。CLL向多形性大细胞淋巴瘤转化者称Richter综合征,发生率约3%~15%。
4.免疫学检查
利用流式细胞仪可以检测细胞表面分化抗原、膜表面免疫球蛋白(SIg)和κ、λ轻链,以确定细胞是否是克隆性增殖并提供进一步分型。典型的慢淋细胞表型为 CD5 + 、CD10 - 、CD19 + 、CD20(dull)、CD23 + 、CD103 - 、FMC7 - ,B细胞慢淋膜表面的免疫球蛋白密度较低,但具有大量胞质免疫球蛋白,CD22、CD79b的表达很弱或缺失。大约50%CLL患者表达CD38(>30%),CD38 + CLL细胞无 IGHV 基因突变,与ZAP-70同为慢淋预后指标。
50%~75%的患者有低γ球蛋白血症,以IgM减少为著,少数为无丙种球蛋白血症。5%的患者可出现单克隆免疫球蛋白血症,一旦IgM明显增高,则临床表现类似巨球蛋白血症。少数患者可出现μ重链病或轻链型蛋白尿。20%~30%的患者直接Coombs试验阳性。
5.染色体和基因检查
大约50%的患者有染色体数目及结构异常,多为11、12、14和13号染色体异常,常见的染色体畸变有del(11q)、del(13q)、+12、del(17p)等。基因突变可涉及 p53 、 NOTCH1 、 SF3B1 、 BIRC3 、 MYD88 等,其结果有助于治疗和预后分层。
【诊断与鉴别诊断】
从年龄、临床表现、外周血白细胞>10×10 9 /L、淋巴细胞比例≥50%、淋巴细胞绝对值>5×10 9 /L、骨髓象淋巴细胞>40%且以成熟淋巴细胞为主以及淋巴结肿大等典型表现,多数病例诊断并不难。持续性淋巴细胞增多最具诊断意义。有淋巴结肿大须与淋巴结结核、淋巴瘤及慢性炎症所致的淋巴结病变相鉴别。淋巴细胞增多者应区别于传染性单核细胞增多症、麻疹、水痘、巨细胞病毒感染等反应性淋巴细胞增多。其他慢性淋巴增殖性疾病如幼淋巴细胞白血病、毛细胞白血病、各种类型淋巴瘤,如小淋巴细胞淋巴瘤、套细胞淋巴瘤、脾边缘区淋巴瘤、滤泡中心性淋巴瘤等,流式细胞仪检测细胞表面抗原有助于各种疾病之间的鉴别(图16-3-3)。
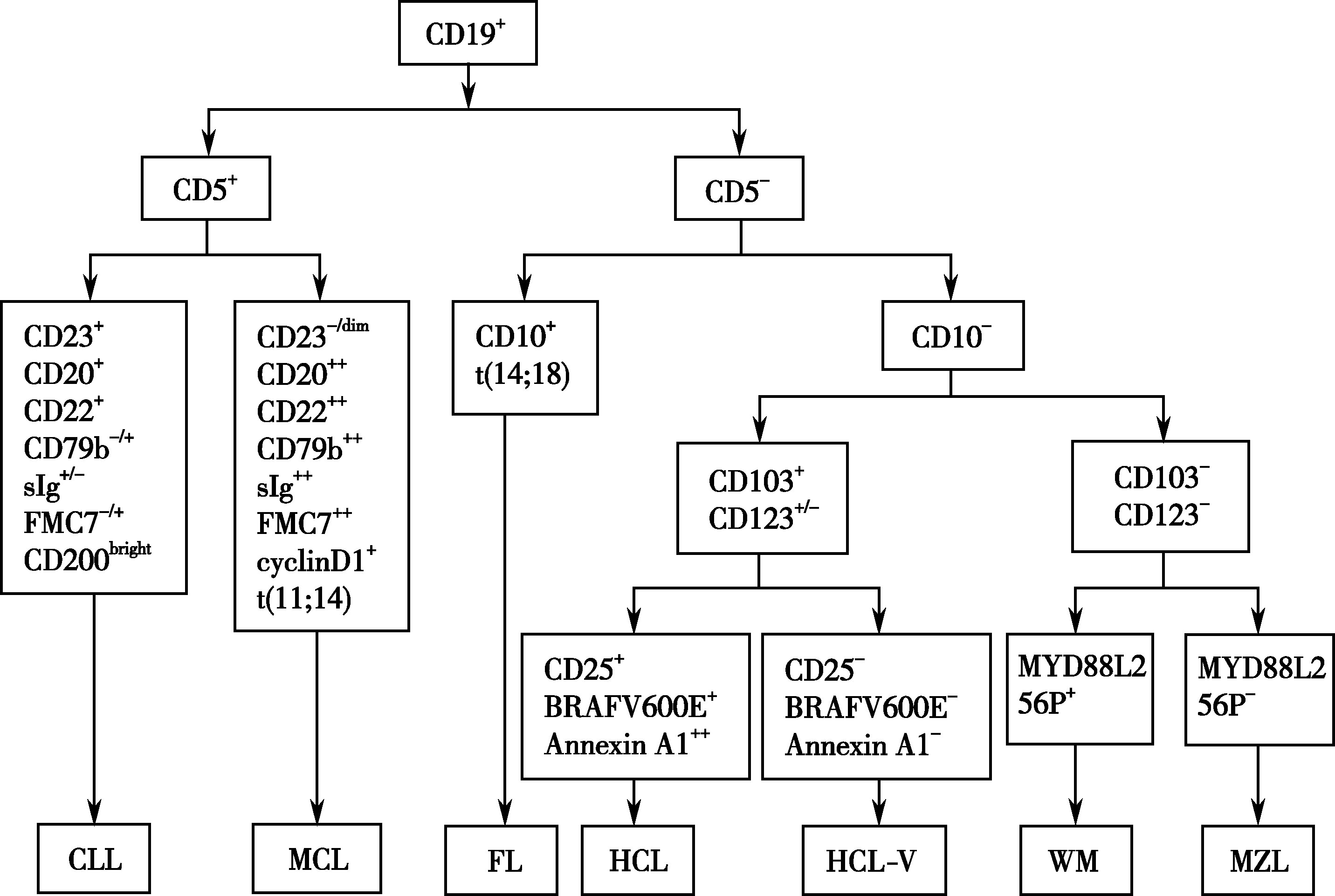
图16-3-3 B细胞慢性淋巴增殖性疾病鉴别诊断流程
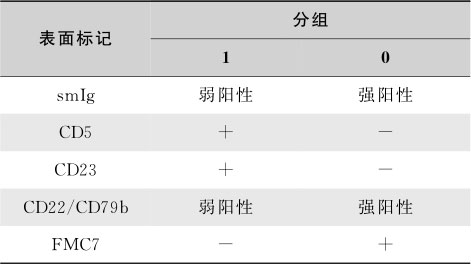
一般慢淋细胞的免疫表型特征为:①表达B细胞分化抗原(CD19、CD20、CD23)和CD5,不表达 T细胞相关抗原;②单克隆表达κ链或γ链;③低表达膜表面免疫球蛋白(smIg)。亦有用CLL诊断评分系统(表16-3-16)与其他B淋巴细胞肿瘤进行鉴别。采用该评分系统,诊断CLL需4~5分,仅少部分CLL为2~3分,其他B细胞淋巴瘤多为1~2分。
2008年WHO分型提出单克隆B淋巴细胞增多症的诊断(monoclonal B-Cell lymphocytosis,MBL),是指免疫表型同CLL,但无淋巴结肿大,外周血淋巴细胞<5×10 9 /L,骨髓淋巴细胞浸润<30%。人群中调查发现>40岁健康人群中3.5%有MBL。MBL的意义不详,有文献称为意义未明单克隆B淋巴细胞增多症(monoclonal B-lymphocytosis of undetermined significance,MLUS),其中有少数 MBL可转变为CLL,在CLL家族中的MBL患者转变率可达13%~18%,但有的MBL患者不转变为CLL。如有明显淋巴结肿大,外周血淋巴细胞<5×10 9 /L,免疫表型同CLL,应诊断为SLL。
表16-3-16 CLL诊断评分系统
【临床分期】
1978年Rai提出的分期法将慢淋分为0~Ⅳ期:0期仅有淋巴细胞增多;Ⅰ期伴有淋巴结肿大;Ⅱ期伴有脾大;Ⅲ期伴有贫血(<110g/L);Ⅳ期伴有血小板减少(<100×10 9 /L)。1987年,Rai将其分期法补充为低危(0期)、中危(Ⅰ、Ⅱ)和高危(Ⅲ、Ⅳ)三组。1981年Binet等提出的分期法共分为3期:A期无贫血(Hb>100g/L)或血小板减少(PLT>100×10 9 /L),肝、脾与颈、腋下、腹股沟淋巴结共5个区域中累及3个以下;B期无贫血或血小板减少,但累及区域≥3个;C期出现贫血和(或)血小板减少。
【治疗】
低危患者或Binet A期淋巴细胞轻度增多(<30×10 9 /L),Hb>120g/L,血小板>100×10 9 /L,骨髓非弥漫性浸润者生存期长,病情稳定者可以定期观察、对症治疗为主。当患者出现发热、体重明显下降、乏力、贫血、血小板降低、巨脾或脾区疼痛、淋巴结肿大且伴有局部症状、白细胞增高且伴有淤滞症状、淋巴细胞倍增时间<6个月、出现幼淋变或Richter变时,应予积极治疗。治疗前应对患者的症状、体征和基因变化进行全面评估,有条件的单位应根据FISH 检查[del(11q)、del(17p)]、患者的年龄和身体状态进行分层治疗。对体能状态好的患者宜选择一线标准治疗,其他患者则使用减低剂量化疗或支持治疗。
(一)化学治疗
1.烷化剂
苯丁酸氮芥(chlorambucil,瘤可宁)为首选药物,完全缓解率为15%,部分缓解率为65%。口服给药剂量为2~4mg/d,可增至6~8mg/d,待淋巴细胞减少50%时减量,稳定后予维持量,也有主张间歇治疗,0.4~0.7mg/kg,第1天口服或分4天口服,每2~4周重复一次。瘤可宁无效者可用环磷酰胺(CTX),口服50~100mg/d,或0.50~0.75g/m 2 静脉注射,每3~4周1次。
2.氟达拉滨(fludarabine)
是单磷酸腺苷氟化物,干扰腺苷代谢,对慢淋有特效。静滴25~30mg/(m 2 ·d),维持30分钟,连用5天,每4周重复1个疗程,初治患者总有效率为70%,完全缓解率达38%,缓解后持续时间较长,近年来有逐渐取代瘤可宁的趋势。主要不良反应有骨髓抑制、免疫抑制持续时间长、神经毒性及易激发自身免疫性溶血性贫血。此外,克拉屈滨(cladribine,2-氯脱氧腺苷)、喷司他丁(pentostatin,2-脱氧助间型霉素)、糖皮质激素以及COP、CHOP、VAD等联合化疗方案等,对慢淋患者有一定疗效。
3.糖皮质激素
糖皮质激素单药治疗对慢淋也有一定疗效,尤其对伴有自身免疫性溶血性贫血或血小板减少的患者较为适用。泼尼松(强的松)40~60mg/d,连用7天,有效后减量并停药,每月重复5天,大约有10%的患者有效。大剂量甲泼尼龙冲击治疗可使部分患者达到部分缓解(PR)的标准,但感染发生的概率也将增大。
(二)放射治疗
有明显淋巴结肿大(包括纵隔或巨脾)、神经侵犯、重要脏器或骨骼浸润且有局部症状者可考虑放射治疗,包括全身放疗(TBI)、全淋巴照射(TNI)和局部照射。与其他方法一起进行序贯治疗可改善全身症状,但持续时间短。放射性核素淋巴结内照射和体外血细胞照射可在一定程度上减少淋巴细胞的数量,但并不延长患者的生存期。
(三)免疫治疗
干扰素-α对早期病例有效,近2/3的患者可获得部分缓解,但对于进展期患者使用干扰素-α可能加速疾病进程。人鼠嵌合的抗CD20单克隆抗体(rituximab,美罗华)对治疗CD20阳性的惰性淋巴肿瘤有特效。Rituximab治疗最常见的不良反应是发热和寒战,少数患者可有溶瘤表现,此外还可能发生恶心、呕吐、高血压或呼吸困难。此外,人源抗CD52单抗CAMPATH-1H可以通过补体依赖细胞毒作用杀灭肿瘤细胞,具有直接抑制淋巴细胞生长的作用,静脉输注或皮下注射均有效。
(四)免疫化疗联合治疗
和传统的单药治疗相比,对CLL患者采用含氟达拉滨的联合治疗方案(FC方案:氟达拉滨20~30mg/m 2 ×3d、环磷酰胺200~300mg/m 2 ×3d;FCR方案:环磷酰胺250mg/m 2 ×3d、rituximab 375~500mg/m 2 ×1d、氟达拉滨 25mg/m 2 ×3d,共治疗6个疗程),可以明显提高初治患者的完全缓解率,但可能增加骨髓抑制和感染等并发症的发生率。
(五)骨髓移植
对年轻、能耐受强烈治疗、具有高危因素(如无 IGHV 基因突变、11q22-q23缺失或17p13缺失)的患者可考虑行骨髓移植。对这些患者主张在早期疾病无进展时进行移植。自体造血干细胞移植可改善患者的无进展生存,但并不延长总生存期,不推荐常规采用。异基因移植具有细胞免疫杀灭肿瘤细胞的优点,是CLL的唯一治愈手段,但移植相关死亡率高于自体移植。
(六)其他治疗
有严重贫血、血小板减少而药物或脾区放疗无效时,可考虑脾切除术;有低γ球蛋白血症、反复感染或自身免疫性疾病者,可定期静脉给予丙种球蛋白(IVIG);淋巴细胞单采可暂时性降低外周血淋巴细胞,减轻器官浸润,增加血红蛋白和血小板数量。
【疗效与预后判断】
患者的症状和体征消失、外周血中性粒细胞>1.5×10 9 /L、血小板>100×10 9 /L、淋巴细胞计数<4×10 9 /L、血红蛋白>110g/L、骨髓中淋巴细胞比例低于30%,且持续2个月以上时认为处于缓解期。如果淋巴细胞绝对计数比起病时减少50%以上、肝脾、淋巴结比起病时缩小50%以上、外周血中性粒细胞>1.5×10 9 /L或比起病时增加50%以上、血红蛋白>110g/L或比起病时增加50%以上、血小板>100×10 9 /L或比起病时增加50%以上,且持续2个月以上,认为是部分缓解。一旦出现新发的淋巴结肿大、上述各项指标比起病时增加50%以上时,则认为疾病进展。
CLL虽发展缓慢,但难以治愈。根据患者性别、起病时的年龄、血清β 2 -微球蛋白(β 2 -MG)、淋巴细胞绝对计数、病情分期、淋巴结区受侵犯的数量可以将患者分为低危(1~3分)、中危(4~7分)、高危(≥8分)三组(表16-3-17)。此外,淋巴细胞倍增时间、 IGHV 基因有无突变、ZAP-70、CD38的表达、骨髓病理分型、染色体核型和乳酸脱氢酶水平等都和预后有关(表16-3-18)。具有 IGHV 基因突变的CLL预后较好,缺乏 IGHV 基因突变的患者生存期短。由于 IGHV 基因测定有一定困难,但可用ZAP-70和CD38作为替代指标。CD38表达和(或)ZAP-70阳性者预后不佳。
表16-3-17 慢淋预后评分
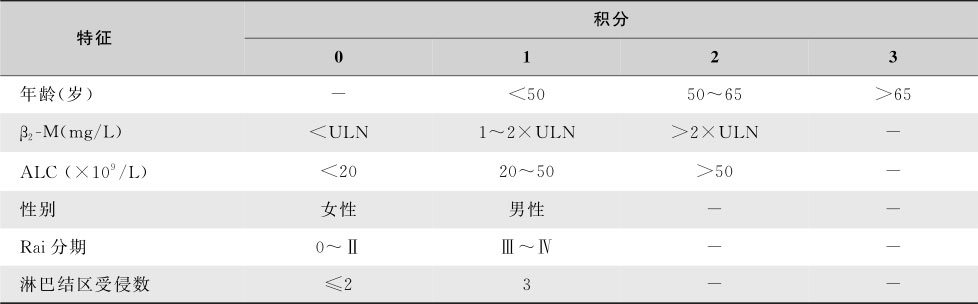
注:ALC:淋巴细胞绝对计数;ULN:正常上限
表16-3-18 慢淋预后指标

续表

患者可向幼淋巴细胞白血病、弥漫大B细胞淋巴瘤(称Richter综合征,Richter syndrome)、霍奇金淋巴瘤、急淋、多发性骨髓瘤等其他恶性淋巴增殖性疾病转化。总之,有下列情况预后不佳:①无免疫球蛋白重链基因重排并表达CD38抗原;②Richter综合征;③幼淋变或混合慢淋/幼淋患者;④急淋变(甚罕见)。CLL中位生存期35~63个月,各期有明显差异,也有患者生存时间长达10年以上。

(资源64) “疑难血液病临床和细胞形态学讨论”之四(病例)
三、其他慢性淋巴增殖性疾病
(一)毛细胞白血病(hairy cell leukemia,HCL)
HCL是一种少见的成熟小B淋巴细胞的惰性肿瘤,诊断时中数年龄约55岁,男∶女=4∶1。几乎所有HCL患者都存在 BRAF V600E 突变,提示 HCL 中存在 RAF-MEK-ERK 这一丝裂原活化蛋白激酶(MAPK)通路的激活,是HCL的发病机制,也是HCL靶向治疗的基础。
经典型HCL的临床特征:全血细胞减少伴单核细胞减少,易发生感染,脾大显著而淋巴结肿大不显著,50%病例骨髓穿刺有干抽(骨髓纤维化)。肿瘤细胞常侵犯骨髓、外周血和脾,具有典型的毛细胞形态(周边有不规则的锯齿状伪足和细长毛发状突起,在电镜下尤为突出),酸性磷酸酶(ACP)阳性,不被酒石酸抑制(TRAP),透射电镜下在胞质内可见核糖体-板层复合物(RLC)。免疫表型具有成熟B细胞标记(CD19 + ,CD20 + ),sIg + ,毛细胞“特异性”标记:抗 BRAF V600E 抗体VE1检测阳性、以及CD103 + 、CD11c + 、CD25 + 、Annexin A1 + 。但HCL-V并不表达 BRAF V600E 。
HCL主要应和绒毛淋巴细胞脾淋巴瘤(splenic lymphoma with circulating villous lymphocytes,SLVL)鉴别,后者肿瘤细胞不表达 BRAF V600E 突变,在细胞两端有短绒毛,借免疫表型亦可鉴别,脾病理HCL主要侵犯红髓,SLVL主要侵犯白髓。
HCL对一般化疗不敏感,切脾仅能缓解脾功能亢进,减少肿瘤负荷。威罗菲尼可以抑制由 BRAF 基因激活而产生的ERK和MEK的磷酸化而使白血病细胞凋亡,在化疗无效的患者中日益得到广泛应用并取得良好的效果。HCL对干扰素-α和核苷类似物[喷司他丁(DCF)和克拉曲滨(2-CDA)]较敏感。复发、难治的患者可联用CD20单抗。病情进展较缓慢,总体10年生存率90%以上。
(二)幼淋巴细胞白血病(prolymphocytic leukemia,PLL)
根据细胞类型分为B-PLL和T-PLL。B-PLL是B幼淋巴细胞侵犯骨髓、外周血和脾所致的恶性肿瘤。诊断时的中数年龄为70岁,男∶女=4∶1。常有巨脾,淋巴结肿大不明显,白细胞计数常>100×10 9 /L,外周血幼淋巴细胞常>55%,有明显的核仁。肿瘤细胞表达较强的膜IgM或IgD以及其他B细胞抗原,表达FMC-7,多数病例CD5 - ;50%的患者有Del(17p)染色体异常。治疗可选用CHOP、氟达拉滨、克拉屈滨、CD20单抗等,中位生存时间仅30~50个月。T-PLL是具有成熟的胸腺后T淋巴细胞表型的幼淋巴细胞增殖所导致的一种侵袭性T淋巴细胞白血病,占PLL病例的1/5,常累及外周血、淋巴结、肝脾和皮肤;表达CD2、CD3和CD7,但不表达 TdT和CD1a;t(14;14)(q11;q32)较多见;中位生存期少于1年。
(三)大颗粒淋巴细胞白血病(large granular lymphocytic leukemia,LGLL)
LGLL可根据细胞类型分为TLGLL和NK-LGLL。2008年WHO分型将T-LGLL单独列出,NK-LGLL归于NK细胞的慢性淋巴增殖性疾病。T-LGLL是一组异质性疾病,以不明原因的外周血大颗粒淋巴细胞持续(>6个月)增高[绝对值(2~20)×10 9 /L)为特征,常侵犯骨髓和外周血。大颗粒淋巴细胞体积较大(红细胞体积×2),胞质丰富,含较多嗜天青颗粒,可粗可细。典型的免疫表型为CD3 + ,CD8 + ,CD16 + ,CD57 + ,TCRαβ + 。患者常出现严重的粒细胞缺乏和贫血,但血小板减少比较少见,严重者可出现纯红细胞再障,常伴有自身免疫性疾病。病情呈惰性,进展缓慢,可予环孢素、CTX、肾上腺糖皮质激素等治疗。
(四)成人T细胞白血病(adult T-cell leukemia/lymphoma,ATLL)
ATLL是一种与人T细胞白血病病毒Ⅰ(HTLV-Ⅰ)感染直接相关、发生于成人的淋巴系统恶性克隆增殖性疾病。具有地域分布的特点,日本西南部、加勒比海地区和中非是高发地点。病变侵犯部位常比较广泛,临床表现多样,可表现为急性型(最常见,以白血病为主要表现,白细胞计数升高)、淋巴瘤型(以显著淋巴结肿大为特征,无外周血受累)、慢性型(常以皮肤肿瘤浸润为主要表现)和冒烟型。肿瘤细胞表达T细胞相关抗原,但CD7阴性,大部分为CD4 + 、CD8 - 。国内诊断标准:①发病于成年人;②有浅表淋巴结肿大,无纵隔和胸腺肿瘤;③外周血白细胞数常增高,淋巴细胞高度多形性,多形核淋巴细胞呈分叶,花瓣样(花细胞)占10%以上,有成熟T细胞表面标志;④血清抗HTLV-Ⅰ抗体阳性。
主要参考文献
1.黄晓军.慢性髓性白血病的过去、现在和未来.中华血液学杂志,2014,35(2):89-91.
2.中华医学会血液学分会,中国抗癌协会血液肿瘤专业委员会.中国B细胞慢性淋巴增殖性疾病诊断专家共识(2014年版).中华血液学杂志,2014,35(4):367-370.
3.中华医学会血液学分会,中国抗癌协会血液肿瘤专业委员会.中国慢性淋巴细胞白血病/小淋巴细胞性淋巴瘤治疗指南.中华血液学杂志,2015,36(10):809-813.
4.Greer JP,Foerster J,Rodgers GM,et al.Wintrobe's Clinical Hematology.12th ed.Philadelphia:Lippincott Williams &Wilkins,2009,2006-2030.
5.Greer JP,Foerster J,Rodgers GM,et al.Wintrobe's Clinical Hematology.12th ed.Philadelphia:Lippincott Williams &Wilkins,2009,2214-2255.
6.Mato A,Porter DL.A drive-through cellular therapy for CLL in 2015:allogeneic cell transplantation and CARs.Blood,2015,126(4):478-485.
7.Kreitman RJ.Removing a hair of doubt about BRAF targeting.Blood,2015,125(8):1199-1200.
第四节
少见类型白血病
许小平
一、急性未定系列白血病
WHO(2001)造血和淋巴组织肿瘤分类将急性未定系列白血病(acute leukemias of ambiguous lineage,ALAL)分为三种亚型:①急性未分化白血病:原始细胞的形态学、细胞化学及免疫表型缺乏足够的证据可划分为髓系或淋系;②急性双系列性白血病:原始细胞同时有粒系和淋系的形态学和(或)免疫表型特征;③急性双表型白血病:原始细胞同时有B系和与T系的形态学和/免疫表型特征。各系列的确定主要参照欧洲白血病免疫分型研究组(EGIL)提出的免疫标记积分系统(表16-3-1),该积分系统规定髓系积分>2分,淋系积分>2分才能诊断ALAL。2008年WHO修订后的造血和淋巴组织肿瘤分类及诊断标准将ALAL分为急性未分化白血病(acute undifferentiated leukemia,AUL)和混合表型急性白血病(mixed phenotype acute leukemias,MPAL)两类,后者又可分为若干亚型。
WHO(2008)修订后的造血和淋巴组织肿瘤分类及诊断标准对于MPAL的各系列细胞的确定依据也重新作了如下说明(表16-3-19)。
表16-3-19 单一原始细胞群超过一个系列的确定条件

续表

ALAL并不多见,据国外文献报道约占所有急性白血病患者的3%以下。
(一)急性未分化白血病
AUL可能起源于造血干细胞阶段,其临床特征与其他类型的急性白血病相似。但原始细胞既不表达cCD79a、cCD22、CD19,也不表达cCD3和MPO,无法归类于髓系或淋系。如检测巨核细胞和浆细胞样树突状细胞的特异性标记亦为阴性。但常可表达HLADR、CD34、CD38及Td T与CD7。诊断AUL须注意排除浆细胞样树突状细胞白血病、NK细胞白血病、急性嗜碱性粒细胞白血病及非造血系统肿瘤。
(二)混合表型急性白血病
1.混合表型急性白血病伴t(9;22)(q34;q11.2); BCR ABL1
该型MPAL的特点是原始细胞存在t(9;22)易位或 BCR-ABL1 重排,是MPAL中最为多见的遗传学异常类型,但总体发病不足所有急性白血病患者的1%。儿童和成人均可发病,但以成人更为多见。由于一些慢粒患者可以进展为混合型原始细胞急变期,此时与本型MPAL诊断标准甚为相符,故特别强调有CML病史患者不应作此诊断。
临床表现与其他类型急性白血病相似。形态学检查很多病例可发现类似于原始淋巴细胞及原始粒细胞的两类原始细胞群,如伴有较显著的成熟阶段髓细胞,应考虑是否为CML急变期。免疫学检测多数患者原始细胞符合B细胞和髓细胞表型,一些病例也可为T细胞和髓系原始细胞。三系列表型的病例也有报道,但甚为罕见。所有患者骨髓细胞染色体核型检查可发现t(9;22)或FISH及PCR检测 BCR-ABL1 融合基因阳性,其中不少患者有复杂染色体核型。治疗可试用伊马替尼及其他酪氨酸激酶抑制剂。
2.混合表型急性白血病伴t(v;11q23);MLL重排
如符合MPAL诊断的患者原始细胞存在涉及 MLL 基因的易位即可诊断本型,但应注意与ALL伴 MLL 基因易位并表达髓系相关抗原的患者相鉴别。本型MPAL可见于儿童和成人,但以儿童更为多见。患者多可发现两种原始细胞群,一群类似与原始单核细胞,另一群类似于原始淋巴细胞。但也有部分病例并无如此显著的形态学特征,只是发现难以分类的原始细胞。免疫分型检测大多数患者可检测到一群CD19 + 、CD10 - 的原始淋巴细胞,CD15常为阳性,CD22和CD79a常弱表达。髓细胞标记应符合MPAL的诊断标准。本型所有患者细胞分子遗传学检测均存在 MLL 基因重排,最常见的伴侣基因是位于4号染色体q21的 AF4。除此之外,t(9;11)和t(11;19)也有报道,但如检测到11q23缺失不应诊断本型MPAL。
3.混合表型急性白血病,B/髓,非特指
MPAL患者如原始细胞符合B细胞和髓系细胞诊断标准,但无前面提及的两类基因异常,即可诊断本型。本型MPAL属罕见类型白血病,大约占所有白血病患者的1%。临床特征与其他类型急性白血病比较无明显特殊之处。原始细胞形态与ALL相似,或存在原始淋巴细胞样和原始粒细胞样两群形态不一致的白血病细胞。免疫表型检测符合上述B淋巴细胞和髓系细胞的诊断标准。MPO阳性的原始粒细胞或原始单核细胞通常也表达其他髓系相关标记如CD13、CD33或CD117,更成熟的B细胞标志如CD20表达较罕见。本型患者大多预后差。
4.混合表型急性白血病,T/髓,非特指
MPAL患者如原始细胞符合T细胞和髓系细胞诊断标准,但无前面提及的两类基因异常,即可诊断本型。本型MPAL较MPAL(B/髓,非特指)更为罕见。可见于儿童与成人,与 MPAL(B/髓,非特指)比较,本型在儿童更为多见。本型 MPAL临床表现与细胞形态学特征均与 MPAL(B/髓,非特指)类似,但原始细胞除表达MPO、CD13、CD33或CD117外,还表达cCD3,CD7、CD5、CD2也常为阳性。如存在起源于T细胞系列的原始细胞群,膜表面CD3亦可阳性表达。细胞遗传学检测结果与 MPAL(B/髓,非特指)类似,即多数病例虽可检测到克隆性细胞遗传学异常,但并无特异性。本型MPAL可试用髓细胞白血病与淋巴细胞白血病相结合的化疗方案治疗,部分病例可能有效,但总体预后不佳。
5.混合表型急性白血病,非特指-罕见类型
混合表型急性白血病,非特指-罕见类型(Mixed phenotype acute leukemia,NOS-rare types)是指白血病患者的原始细胞有明确证据为T和B细胞系列,EGIL双表型白血病的诊断标准(一个以上系列的积分>2),但由于CD79a可以在TALL检测到,因此将CD79a表达定为2分可能会使本型MPAL的诊断过多。CD79a和CD10不应该作为B细胞分化的证据。也会有一些罕见的三系列(B细胞、T细胞及髓系细胞)的病例,但至今未见B或T/巨核细胞白血病以及B或T/红细胞白血病的病例报道。
6.其他未定系列白血病
某些白血病患者虽然表达多系列标志,但既不符合上述 WHO(2008)造血和淋巴组织肿瘤分类标准规定的AUL或MPL的诊断条件,也不能作为单一系列确定。例如原始细胞表达T细胞相关标志CD7和CD5,但不表达T细胞特异抗原胞质CD3,髓系相关抗原CD33和CD13阳性,但不表达MPO。可以暂时将这些病例作为急性不能分型白血病(acute unclassifiable leukemia)考虑。
二、急性全髓增殖症伴骨髓纤维化
急性全髓增殖症伴骨髓纤维化(acute panmyelosis with myelofibrosis,APMF)是 WHO(2001)造血与淋巴组织肿瘤分类方案确定的一种AML亚型。其特征是原发性全髓细胞增生伴骨髓纤维增生。APMF非常少见,主要发生于成年人,也可见于儿童。临床表现主要为全血细胞减少,疲乏无力,无或轻度脾大。病情发展迅速。骨髓穿刺常为“干抽”。骨髓活检有核细胞增多,可见不成熟红系、粒系和巨核细胞均有不同程度增多,原始细胞多数≥20%。巨核细胞增多,而且常有发育不良的形态学改变,网状纤维显著增多,可有胶原纤维但不常见。免疫标记检测原始细胞通常除表达CD34外,还表达一个或多个髓系抗原如CD13、CD33、CD117,MPO一般为阴性。骨髓细胞遗传学检查常为复杂染色体异常核型,多累及5号或(和)7号染色体。多数患者化疗疗效差,生存期通常仅为数月。
三、髓系肉瘤
髓系肉瘤(myeloid sarcoma)曾用的名称有髓外髓系肿瘤、粒细胞肉瘤、原始粒细胞瘤及绿色瘤等,是一种由髓系原始细胞或未成熟髓系细胞形成的肿块,发生于髓外部位或骨骼。最常见的髓系肉瘤类型为粒细胞肉瘤,以往曾根据细胞的成熟程度将其分为3型:①原始细胞型:主要由原始粒细胞组成;②未成熟细胞型:主要由原始粒细胞和早幼粒细胞组成;③成熟细胞型:主要由早幼粒细胞和偏成熟的中幼粒细胞组成。现在认为这种区分意义不大。另一种较少见的髓系肉瘤为原始单核细胞肉瘤,主要细胞构成成分为原始单核细胞。慢性骨髓增殖性疾病的急性转化期,可出现粒、红、巨核3系造血细胞增殖的髓系肉瘤,肉瘤的细胞成分也可以红系前体细胞或巨核细胞增殖为主。骨髓增生异常综合征(MDS)患者亦有发生髓系肉瘤的报道。
髓系肉瘤常发生于儿童及青年,男多于女。肿块可为单个、多个或播散性。可单独出现或与AML、MPD及MDS伴发。常见的发生部位为颅骨、副鼻窦、胸骨、肋骨、椎骨、盆骨的骨膜下,淋巴结及皮肤也较常见,乳腺、肝、脾、肾、肌肉偶有累及。髓系肉瘤发生于眼眶骨膜下,可引起突眼症。以一侧或双侧不对称的突眼最为典型。
髓系肉瘤易与淋巴瘤混淆,鉴别主要依靠细胞化学染色和免疫表型检测。细胞化学染色原始粒细胞及中性粒细胞MPO与氯乙酸ASD萘酚酯酶阳性,原始单核细胞非特异性酯酶阳性。免疫表型检测显示大部分髓系肉瘤CD43阳性。原始粒细胞表达髓系相关抗原CD13、CD33、CD117、MPO。原始单核细胞CD14、CD116、CD11C阳性,溶菌酶及CD68亦可为阳性。治疗一般采用AML相似的化疗方案。放疗对仅有局部瘤块而无白血病表现者有较好的治疗效果,但单独应用认为不能阻止其最终向白血病阶段发展。

(资源65) “疑难血液病临床和细胞形态学讨论”之五(病例)
四、急性嗜碱性粒细胞白血病
急性嗜碱性粒细胞白血病(acute basophilic leukemia,ABL)占AML的1%以下,可见于各年龄组。由于嗜碱粒细胞颗粒中含组胺和肝素,患者的临床表现除一般急性白血病的症状外,还可出现高组胺血症的症状,如皮肤瘙痒、水肿、荨麻疹样皮疹、心动过速、哮喘及溃疡等;肝素释放过多可导致凝血功能异常。本病的外周血和骨髓中的原始细胞形态为中等大小,核浆比例高,核呈卵圆形、圆形或双叶型,有1~3个明显核仁。胞质中度嗜碱性,含数量不等的粗嗜碱性颗粒。电镜下可见幼稚嗜碱性粒细胞或肥大细胞中有特征性的θ颗粒。细胞化学最典型的特征是甲苯胺蓝异染性。原始细胞酸性磷酸酶呈弥漫性阳性,PAS块状阳性,SBB、MPO、NSE为阴性。免疫表型检查原始细胞表达髓系抗原如CD13、CD33及早期造血细胞标记CD34、HLA-DR。CD9、CD22和TdT也可阳性。但特异性淋系相关抗原阴性。表达CD203c,不表达CD117对ABL诊断很有帮助。治疗可采用AML的化疗方案,但预后很差。
五、急性原始巨核细胞白血病
急性原始巨核细胞白血病(acute megakaryoblastic leukemia,AMBL)即 FAB分型中的 M 7 ,约占 AML的3%~5%。临床表现与其他类型AML相似,2/3病例有血细胞减少。血小板大多减少,但也有增多的病例。脾脏肿大并不常见。男性年轻患者,常可与纵隔生殖细胞肿瘤同时存在。诊断主要依靠骨髓检查,骨髓中≥50%为巨核细胞,其中原始细胞≥20%。外周血可有小巨核细胞,原始巨核细胞碎片、异常大的血小板和颗粒少的中性粒细胞。常可伴骨髓纤维化。骨髓活检既可见分化不良的原始巨核细胞单一增多,也可见分化不良的原始巨核细胞与发育异常的成熟巨核细胞混合增生;网状纤维可有不同程度的增多。原始巨核细胞表达一种或多种血小板糖蛋白,如CD41(GPⅡB/Ⅲa)和(或)CD61(GPⅢa),偏成熟型血小板相关标记CD42(GPⅠB)很少表达。原始巨核细胞MPO及其他髓系标记阴性,不表达淋系及Td T,可异常表达CD7。本病的预后较差。
六、自然杀伤(NK)细胞白血病
(一)慢性NK细胞淋巴增生症
慢性NK细胞淋巴增生症(chronic lymphoproliferative disorders of NK cells,CLPD-NK)是WHO(2008)造血与淋巴组织肿瘤分类标准中新列的疾病实体。CLPD-NK约占所有大颗粒淋巴细胞疾病的5%左右。其特征是在无明确病因的情况下外周血NK细胞≥2×10 9 /L持续6个月以上,而这些增殖的NK细胞是肿瘤性的还是反应性的目前仍无法确定。
患者发病无明显性别差异,中位年龄为60岁。病变主要累及外周血和骨髓。多数患者临床表现为外周血大颗粒淋巴细胞计数持续增高而不伴有发热、肝脾大以及淋巴结肿大。部分患者可有血细胞减少,以中性粒细胞减少和贫血为多见。淋巴结肿大、肝和脾大以及皮肤损害少见。CLPD-NK也可与实体瘤、造血系统肿瘤、脾切除、神经病变和自身免疫性疾病并存。
CLPD-NK患者的外周血NK细胞形态中等大小,核仁圆,染色质凝集,胞质轻度嗜碱性,含有细致或粗糙的嗜天青颗粒。免疫学标记检测膜表面CD3阴性,但细胞质CD3ε常为阳性,CD16阳性,CD56常为弱表达,细胞毒标志TIA1、粒酶B、粒酶M阳性。CD2、CD7、CD57减弱甚至消失。
目前国内外尚无CLPD-NK的统一诊断标准,日本学者Oshimi提出只要外周血sCD3 - CD56 +/- CD16 + 的NK细胞>0.6×10 9 /L,持续时间存在超过6个月即可作出诊断。
患者一般临床呈惰性过程,但也有一些患者在疾病进展过程中出现淋巴细胞增生和血细胞减少加重。少数转化为侵袭性NK细胞疾病的病例多见于慢性活动性EB病毒感染者。CLPD-NK的治疗尚无临床试验报告可供借鉴,免疫抑制治疗可作为一线推荐。
(二)侵袭性NK细胞白血病
侵袭性NK细胞白血病(aggressive NK-cell leukemia,ANKL),又称为 NK细胞大颗粒淋巴细胞白血病。是一种sCD3 - 的肿瘤,大约占所有大颗粒淋巴细胞增殖性疾病的10%左右。
ANKL在亚洲地区相对较多见,发病因素可能与EB病毒感染有关。患者多为青少年,中位年龄在39~42岁之间,男女发病大致相当。患者常表现为发热、全身症状及白血病血象,外周血中白血病细胞比例高低不一,少数可高达80%以上。贫血、中性粒细胞减少、血小板减少较为常见。肝、脾常肿大,全身淋巴结亦可肿大,但皮肤病变较为少见。患者可合并凝血异常和噬血细胞综合征。少数病例可能系结外NK/T细胞淋巴瘤或由CLPD-NK转变而来。实验室检查白血病细胞比正常大颗粒淋巴细胞略大,核仁清楚或不明显,胞质丰富,淡染或略呈嗜碱性,含细致或粗糙的嗜天青颗粒。噬血现象易见。免疫表型检测白血病细胞呈CD2 + ,胞膜CD3 - ,胞质CD3ε、CD56和细胞毒性分子阳性,CD11b和CD16也常阳性,而CD57通常阴性。如CD94阳性,提示细胞起源于成熟阶段。
多数患者呈侵袭性、暴发性的临床过程。采用中高度恶性NHL化疗方案疗效常不佳,患者多在几周至6个月内死亡。
(三)NK淋巴母细胞白血病/淋巴瘤
NK细胞淋巴母细胞白血病/淋巴瘤(natural killer cell lymphoblastic leukemia/lymphoma)是 WHO(2008)分类中新列的一类造血与淋巴组织肿瘤,主要指以往文献报道中常以“髓系/NK前体细胞急性白血病(myeloid/natural killer cell precursor acute leukemia)”命名的一类疾病,髓/NK细胞急性白血病(myeloid/natural Killer cell acute leukemia)可能也属其中。这两种疾病的免疫表型的共同特点是均表达CD56,髓系抗原和T细胞相关抗原程度不定表达。多数学者认为肿瘤细胞起源于NK前体细胞,但由于缺乏足够的证据,WHO(2008)分类暂将它们归属于系列未明的急性白血病(acute leukemia of ambiguous lineage)大类中,并且建议今后应对这类疾病检测NK细胞更具特异性的标记如CD91、CD161及CD94 1A转录本、抗KIR抗体谱以明确其起源。
诊断NK淋巴母细胞白血病/淋巴瘤应与母细胞性浆细胞样树突状细胞白血病仔细鉴别。
(四)母细胞性浆细胞样树突状细胞肿瘤
1994年Adachi等首先报道了1例肿瘤细胞具有淋巴母细胞样形态特点并且表达CD4和CD56的患者。至1998年,类似的病例共有14例报道。由于肿瘤细胞表达CD56,缺乏T和B细胞标记及髓系特异性抗原,当时推测疾病起源于NK细胞。1999年,Petrella等发表了当时病例数最多(7例)的一组研究报告,提出这是不同于其他类型淋巴瘤的一种疾病实体。WHO(2001)分类暂将其命名为母细胞性NK细胞淋巴瘤(blastic NK cell lymphoma)。由于CD56阳性并不是NK细胞的特异性标记,CD4阳性亦非NK细胞发育过程中典型表达,不少学者对其疾病起源于NK细胞的母细胞阶段的观点不断提出质疑。2001年与2002年,Chaperot等和petrella等分别报告肿瘤细胞表面CD123强阳性表达,这一发现对研究肿瘤细胞的起源提供了重要的线索,因为CD123主要表达于树突状细胞(DCs)和浆细胞样树突状细胞(plasmacytoid dendritic cells,pDCs)。WHO(2008)分类已将疾病名称改为母细胞性浆细胞样树突状细胞肿瘤(blastic plasmacytoid dendritic cell neoplasm,BPDCN),并将其归入“急性髓细胞白血病和相关髓系肿瘤”大类之中。
本病罕见,占急性白血病的比例<1%。多见于老年人,诊断时中位年龄66岁,其中女性患者55.5岁,男性患者67.0岁。男女比例为3.5∶1。BPDCN的临床特征及进展模式主要有两种类型:一型(90%患者)主要以惰性皮肤病变开始,随后发生肿瘤播散;另一型(10%患者)起病时即累及全身,表现为白细胞计数增高,骨髓大量原始细胞浸润等急性白血病特征,该型患者可伴有皮肤多发性结节。
多数患者常表现为无症状性皮肤损害,皮肤病变可表现为结节、斑块或类似挫伤。有些患者表现单一皮肤病变,而更多的患者则表现为多发性病变,可发生在身体任何部位。病变可表现为皮肤红斑,呈淡红色或淡蓝色;直径从数毫米到数厘米不等。20%患者有局部或播散性淋巴结增大,40%~60%的患者有脾脏增大。40%~90%的患者起病时即有骨髓累及,但因数量少易被忽视。随着病情进展,骨髓肿瘤细胞逐渐增多,并出现贫血和血小板减少。这些患者甚至被诊断为骨髓增生异常综合征(MDS)与本病共存。在疾病的终末期或复发阶段常有急性白血病的表现。骨髓检查见单一形态的肿瘤细胞弥漫型浸润,细胞化学染色MPO与酯酶阴性。超过50%的患者表达TdT,但细胞核TdT的染色在不同部位的肿瘤组织差异较大,如淋巴结要高于皮肤。有人认为肿瘤细胞如表达EBV抗原、MPO、溶菌酶、PAX-5、CD20、CD22和T细胞受体蛋白应排除本病的诊断。
细胞遗传学检查66%的BPDCN患者具有异常核型;未发现特异性染色体畸变,但一个显著的特征是在同一个肿瘤细胞内可发生平均6~8种染色体异常。最常见的为5q缺失(72%),其他如13q、12p、6q改变和15号、9号丢失也较易见,8号和21号三体以及7号单体较为罕见。
疾病进展迅速,预后较差。临床通常采用急性白血病的诱导方案,初次治疗患者80%~90%对联合化疗有效,但多数患者在短期内复发并产生耐药,中位存活时间为12~14个月。对于年轻患者应在首次缓解后及时行异基因造血干细胞移植。
国外文献还报道少数肿瘤细胞为母细胞形态、免疫表型特征为CD4 - 、CD56 + 、CD13 - CD33 - 的患者,这些患者大多CD3、CD19、CD20和 MPO表达缺失。对于这部分患者的疾病分类目前存在较大争议。一些学者认为这部分患者属于BPDC变异型。
七、低增生性急性白血病
低增生性急性白血病目前并不认为是一种独立的疾病实体,可能是AML患者起病时的一种比较少见的疾病状态。发生率大约占AML的2%~10%。男性多于女性,半数以上年龄大于50岁。大多起病隐袭,病程缓慢,白血病细胞浸润现象不明显,肝、脾、淋巴结多不肿大。外周血三系列细胞减少,骨髓增生减低,多部位骨髓活检示造血组织≤50%,但原始细胞≥20%。细胞类型以粒细胞为多,也可单核细胞或粒、单细胞混合型。化疗缓解后骨髓增生活跃,病情复发时增生又呈低下。
由于患者多为老年人,大剂量化疗容易导致造血功能衰竭,故多主张先以支持疗法,待病情稳定后再予小剂量阿糖胞苷、阿克拉霉素或三尖杉碱间歇静滴。也有报道用标准剂量化疗可获得较好疗效。
主要参考文献
1.Jaffe ES,Harris NL,Vardiman JW,et al.血液病理学.陈刚,李小秋译.北京:北京科技出版社,2013,743-912.
2.Swerdlow SH,Campo E,Harris NL,et al.WHO Classification of Tumors of Haematopoietic and Lymphoid Tissues.4th ed.Lyon:IARC,2008,130-284.
3.Swerdlow SH,Campo E,Harris NL,et al.WHO Classification of Tumors of Haematopoietic and Lymphoid Tissues.4th ed.Lyon:IARC,2008,150-155.
第五节
骨髓增生异常综合征
邹善华
骨髓增生异常综合征(myelodysplastic syndrome,MDS)是一组造血干/祖细胞获得性克隆性疾病,伴三系血细胞发育异常(又称病态造血),进行性、难治性外周血细胞减少,骨髓无效性造血;具有向AML转化的高危险性。

(资源66) “疑难血液病临床和细胞形态学讨论”之六(病例)
【病因与发病机制】
原发性MDS病因不明,继发性MDS与接触放射线、苯或接受烷化剂、拓扑异构酶Ⅱ抑制剂类化疗药物治疗有关。治疗后发生的 MDS称治疗相关性 MDS(therapy-related MDS,t-MDS)。
MDS的发病机制尚未完全明确。通过大量研究证实MDS是源于骨髓造血干/祖细胞的克隆性疾病。在 MDS的发生发展过程中,包含了骨髓细胞的凋亡、增殖及克隆扩张等多重机制。用单克隆抗体免疫标记三系细胞,分别测定凋亡率,结果发现,无论是总的凋亡率还是红系、粒系、巨核系的凋亡率均明显增高。Fas-FasL系统及肿瘤坏死因子-α(TNF-α)在骨髓细胞凋亡过程中起重要作用。在MDS进展为AML的过程中凋亡的功能则逐渐丧失。在MDS的患者中,细胞遗传学异常较为常见,如5q-、+8、-7等,治疗相关性MDS多为复合染色体异常。表观遗传学的改变在MDS的发病中起重要作用,涉及RNA剪接、DNA甲基化、组蛋白修饰、转录、DNA修复调控、黏结蛋白、RAS通路、DNA复制等多方面的基因突变,造成DNA的高度甲基化和组蛋白去乙酰化等现象。免疫机制异常在MDS中的发病意义目前亦得到重视。研究发现体液免疫和细胞免疫均可受累。部分患者可产生异源抗体或自身抗体。MDS患者体内存在T细胞的克隆性扩增,效应性CD8 + T淋巴细胞数量增多,且T细胞受体Vβ有显著增多的异常Vβ谱系。另外血管内皮生长因子(VEGF)的分泌增多,骨髓微小血管密度明显增高,在 MDS的发病中也有一定意义。
【分型】
(一)FAB分型
FAB协作组于1982年根据血液学的特点将MDS分为5型(表16-3-20)。
表16-3-20 FAB协作组的分型标准(1982)
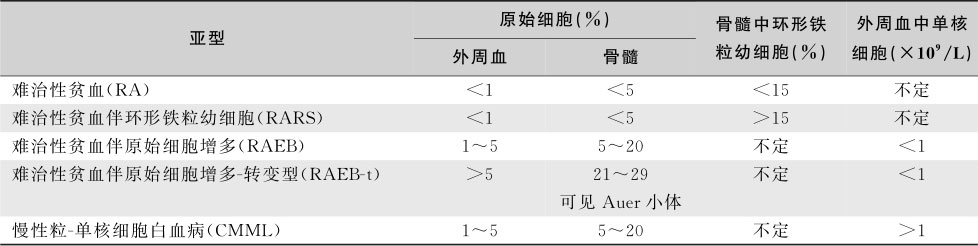
FAB分型中,RAEB及RA所占比例较高,但CMML国内较国外少见。FAB分型有助于判断预后。RA、RAS部分可转变为RAEB、RAEB-t或原始细胞<5%CMML,后者又有部分病例会转变为急性白血病。
(二)WHO分类
FAB分型有许多优点,使用方便,但亦存在不足之处。WHO 2001年分类结合了细胞遗传学的分析,进一步完善了MDS的分类。2008年又进行了若干补充和修改(表16-3-21)。WHO分类建议,只要有单系病态造血并排除其他原因血细胞减少至少持续6个月,即可诊断难治性血细胞减少伴单系发育异常(refractory cytopenia with unilineage dysplasia,RCUD),包括难治性贫血(refractory anemia,RA),难 治性中 性粒 细胞减 少(refractory neutropenia,RN)和难治性血小板减少(refractory thrombocytopenia,RT)。难治性贫血伴环状铁粒幼细胞(refractory anemia with ring sideroblasts)也仅有红系发育异常。据骨髓中原始细胞比例的多少将难治性贫血伴原始细胞增多(refractory anemia with excess of blasts,RAEB)分为1型(骨髓原始细胞<10%)和2型(原始细胞≥10%);较FAB分类增加了难治性血细胞减少伴多系增生异常(refractory cytopenia with multilineage dysplasia,RCMD),5q-综合征和不能分类 MDS。将FAB分型中属于CMML的患者归MDS/MPN;AML诊断标准修定为原始细胞≥20%,把RAEB-t从MDS中剔除。原FAB分型诊断的RAEB-t是否应按AML治疗需要取决于患者个体情况而定。
表16-3-21 WHO的MDS分类及其标准

续表
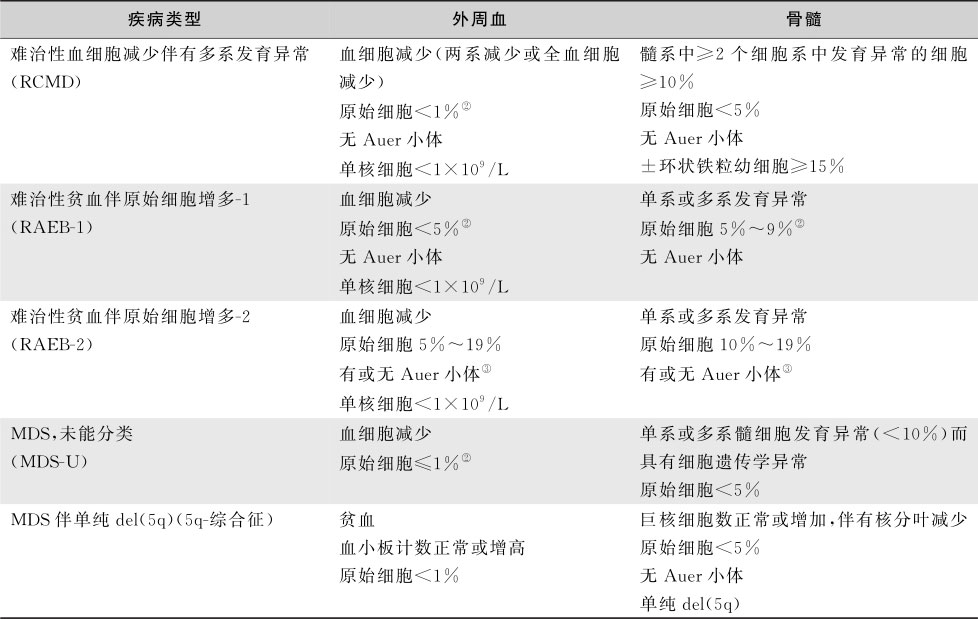
注:①RCUD如伴全血细胞减少,应诊断为MDS-U;②如骨髓原始细胞<5%,外周血为2%~4%,应诊断为RAEB-1;RCUD和RCMD外周血原始细胞1%,应诊断为MDS-U;③伴Auer小体,而外周血原始细胞<5%,骨髓原始细胞<10%,应诊断为RAEB-2
【临床表现】
患者以50岁以上中老年人居多。国外报道中青年及儿童极少见。儿童MDS十分罕见,据统计<14岁儿童MDS仅占该年龄组造血系统肿瘤患者5%以下。儿童MDS具有一定特点,如骨髓低增生甚常见,5q - 综合征和RARS甚罕见。因此,2008年 WHO分类将儿童MDS另列一个亚型。据国内报道中青年患者并不十分少见。男性多于女性。多数患者起病隐匿,可无症状。85%以上的患者有贫血,如患者贫血较严重,可有头昏、乏力、全身不适、活动后心悸或气促等症状。这种贫血可能在就诊前已存在数年,体检时才被发现。亦可有体重下降。原因不明的发热占10%~15%,多数为低热,与感染无关。高热多数由于白细胞减少,并发感染引起。1/3病例可因血小板减少出血,包括皮肤瘀点瘀斑,牙龈出血,轻微损伤后发生血肿。如起病时即有严重的白细胞及血小板减少,则预示预后不良。少数患者可有关节疼痛或类似结缔组织病的症状,极少数病例可发生发热性中性粒细胞性皮炎(Sweet综合征)或坏死性脓皮病(pyoderma gangrenosum)。肝脾大,多数为轻度,见于5%~20%病例。淋巴结一般不肿大。本病转化为急性白血病后,病程短促,疗效甚差。
【实验室检查】
(一)血常规及骨髓涂片
血常规及骨髓象检查显示血细胞形态和数量的异常变化是诊断的重要依据之一。血常规检查有全血细胞减少的病例占半数以上,部分病例仅为一系或两系血细胞减少。一系减少常为红细胞减少,并可能在全血细胞减少前存在数年。大多数病例骨髓造血细胞呈显著增生。部分病例为增生或增生活跃,仅极少数病例增生低下。增生低下者可称低增生性MDS,仅占10%,但无独立预后意义。90%以上的病例有不同程度的细胞发育异常(病态造血)。各系细胞形态的变化归纳如下:
1.红细胞系
骨髓中幼红细胞可过多(>60%)或过少(<5%)。红系病态造血主要表现在核的改变,包括核出芽、核间桥、核碎裂、多核和巨幼样变。胞质改变包括特征的环状铁粒幼细胞(含5个以上粗大的铁蛋白颗粒,且围绕核周1/3以上)、空泡及PAS阳性。血常规示成熟红细胞大小和形态不一,可有巨大红细胞、椭圆形红细胞及有核红细胞,染色过浅和嗜点彩红细胞。其中以奇数核和巨大红细胞最有诊断意义。
2.粒细胞系
骨髓中原始细胞比例增加,胞体大小不一,核分叶过多或过少,呈假性Pelger-Hüet畸形,胞质嗜碱性强,颗粒可减少或缺乏并分布不均匀,呈假性Chediak-Higashi颗粒及Auer小体等。血常规有幼稚粒细胞出现和单核细胞增多。其中原始细胞可分为2型:Ⅰ型为无嗜天青颗粒的原始细胞;Ⅱ型为含有嗜天青颗粒但未出现核旁高尔基区的原始细胞,出现核旁高尔基区者则判断为早幼粒细胞。
3.巨核细胞
骨髓中出现淋巴样小巨核细胞、单圆核或多圆核巨核细胞。血片中可有巨大或畸形的血小板。其中以淋巴样小巨核细胞最有诊断意义。
4.细胞化学
半数患者骨髓部分早幼粒细胞和其他不同发育阶段中性粒细胞过氧化酶活性可降低,约20%患者的中性粒细胞碱性磷酸酶积分下降。幼红细胞糖原染色(PAS)可阳性。骨髓中除出现环状铁粒幼细胞外,贮存铁亦增加,幼稚粒单细胞α萘酚醋酸脂酶及氯醋酸脂酶可呈双重阳性。
(二)骨髓活检
与骨髓穿刺涂片相比,对估计骨髓内细胞的数量,增生程度更准确,也可以观察到骨髓内的组织结构、造血细胞的形态及其分布。对骨髓涂片质量不满意或骨髓穿刺出现“干抽”时,诊断的意义更大。通常在髂后上棘取骨髓组织,长度不少于1.5 cm。骨髓活检组织中可见到原粒细胞及早幼粒细胞在小梁旁区或小梁间中央区形成集丛(3~5个细胞)或集簇(>5个细胞),称为“幼稚前体细胞异常定位”(abnormal localization of immature precursors,ALIP),每张骨髓切片上都能看到至少3个集丛或集簇为ALIP(+)。对诊断有特殊的意义,也有人认为出现ALIP的病例预后差。切片中亦可见到幼红细胞聚集成堆,原红细胞增多,伴成熟障碍。小巨核细胞增多。怀疑为 MDS的患者建议进行Gomori银染色和原位免疫组化(immunohistochemical,IHC),常用的 检测 标志包括 CD34、MPO、GPA、CD61、CD42、CD68、CD20和 CD3。部分患者伴骨髓网硬蛋白纤维增生。有显著骨髓纤维化约占10%MDS病例,称MDS伴骨髓纤维化,多表示疾病进入进展期,有原始细胞增多。
(三)骨髓细胞培养
大多数病例细胞培养的生长不正常,尤其是RAEB及RAEB-t的病例。粒-巨噬系集落形成单位(GM-CFU)集落的生长减少,集簇增多,细胞有成熟障碍。细胞培养生长对预后判断有意义。生长方式可分为两类:一类为非白血病性,患者的中数生存期长、转化为急性白血病的百分比低;另一类为白血病性,患者的中数生存期短,转变为急性白血病的比例高。此外,也有人认为,根据细胞培养生长的情况及造血因子对细胞生长及分化的刺激作用,可使今后对本病的治疗更具有针对性。
(四)骨髓染色体检查
40%~60%病例有非随机的染色体异常,在继发性的MDS中,比例更高,可达80%左右。常见的变化包括染色体全部或部分缺失、染色体数目增多,但易位少见。染色体的异常在RAEB及RAEB-t型多见,在RA及RAS型少见。常见的染色体变化为-5,5q-,-7,-7q-,+8,20q-和-Y等。MDS患者常见的染色体异常中,部分异常具有特异性诊断价值,包括-7/7q-、5/5q-、i(17q)/t(17p)、-13/13q-、11q-、12p-/t(12p)、9q-、idic(X)(q13)、t(11;16)(q23;p13.3)、t(3;21)(q26.2;q22.1)、t(1;3)(p36.3;q21.2)、t(2;11)(p21;q23)、inv(3)(q21;q26.2)和t(6;9)(p23;q34)。5q-综合征常有明显的大细胞性贫血,血小板通常正常或增多,骨髓增生正常或明显增生,巨核细胞的核分叶较少。在临床上以中老年女性多见,中位年龄65岁,多数为输血依赖的RA,病情常较稳定,发生急性白血病的较少。+8、20q-和-Y亦可见于再生障碍性贫血及其他非克隆性血细胞减少疾病,部分伴有单纯+8、20q-或-Y的患者免疫抑制治疗有效,且长期随访未出现提示MDS的形态学依据。染色体的变化对随访病情,估计预后有参考意义。有染色体异常的病例较染色体正常的预后差。在非整倍体的病例中,亚二倍体比超二倍体的预后差。对初诊时核型正常而在随访中出现染色体异常者,预后差,预示有转化为急性白血病的征兆,对有复杂染色体异常者尤为如此。形态学未达到标准(一系或多系细胞发育异常比例<10%)、但同时伴有持续性血细胞减少的患者,如检出具有MDS诊断价值的细胞遗传学异常,应诊断为MDS不能分类(MDS-U)。为提高部分MDS患者细胞遗传学异常检出率,对疑似MDS者,遇到骨髓干抽、无中期分裂相、分裂相质量差或可分析中期分裂相<20个时,可进行FISH检测,通常探针应包括:5q31、CEP7、7q31、CEP8、20q、CEPY和p53。
(五)免疫学检查
本病患者免疫学检查可异常。许多患者可有多克隆性高γ球蛋白血症,最多见于CMML型,在RAS型,也可有低γ球蛋白血症,约12%病例可有单克隆球蛋白。血液中的淋巴细胞减少,主要是CD4 + 细胞的减少,CD4/CD8比值降低。也有证据表明T细胞有克隆性增殖。T细胞的功能也可异常,对植物血凝素(PhA)和刀豆球蛋白(ConA)刺激,淋巴细胞的增殖反应降低。自然杀伤细胞活性降低,并伴随出现对干扰素反应的丧失或干扰素产生的不足。许多自然杀伤细胞的活性经过与α干扰素孵育培养后可恢复。MDS中,单核细胞及巨噬细胞的功能也有异常。部分患者可出现抗核抗体、类风湿因子、Coombs试验阳性。
(六)流式细胞术检测
目前尚未发现MDS特异性的抗原标志或标志组合,但流式细胞术对于低危MDS与非克隆性血细胞减少症的鉴别诊断有应用价值。对于无典型形态和细胞遗传学证据、无法确诊MDS的患者,若流式细胞术检测有≥3个异常抗原标志,可提示MDS存在的可能。
(七)分子遗传学检测
单核苷酸多态性微阵列(SNP-array)等基因芯片技术可以在多数MDS患者中检测出DNA拷贝数异常和单亲二倍体,从而进一步提高MDS患者细胞遗传学异常的检出率。若有条件,SNP-array可作为常规核型分析的有益补充。随着基因芯片、第二代基因测序等高通量技术的广泛应用,多数MDS患者中可检出体细胞性基因突变,常见突变包括 TET2 、 RUNX1 、 ASXL1 、 DNMT3A 、 EZH2 、 N-RAS / K-RAS 、 SF3B1 等。对常见基因突变进行检测对于MDS的诊断有潜在的应用价值。
【诊断和鉴别诊断】
(一)诊断
临床上出现贫血,和(或)伴有感染、出血;外周血有一系、二系或全血细胞减少,外周血有巨大红细胞、巨大血小板、有核红细胞等情况;骨髓为增生性骨髓象,红系比例明显增加,有一系或两系甚至三系血细胞的发育异常(病态造血);骨髓活检有ALIP现象,或伴有纤维组织增生;染色体核型异常。除外其他伴有病态造血的疾病、其他全血细胞减少的疾病,其他红系增生性疾病,可考虑MDS,再进一步分型。在MDS的诊断和分型中,血片和骨髓涂片质量非常重要,应采用新鲜标本,加抗凝剂者应在2小时内进行检查,外周血分类应计数200个白细胞,骨髓要计数500个有核细胞。由于骨髓血细胞病态造血并无特异性,许多非克隆性疾病包括遗传性、营养性、药物性、感染性、中毒性疾病等都可出现血细胞病态造血,因此要注意血细胞病态造血出现的质与量,WHO规定髓系病态造血改变血细胞应≥10%才有意义。
2007年出台了MDS维也纳最低诊断标准(表16-3-22),诊断MDS需具备两个必要条件和至少一条决定性标准,对于符合必要条件、不符合决定性标准(如非典型染色体核型异常,病态造血细胞<10%,原始细胞占4%等),但又具有典型MDS相关的临床表现(如输血依赖的大细胞性贫血)的患者,应进行其他检查(辅助诊断标准),以得出诊断或进行随访(部分可暂时归为意义未明的特发性血细胞减少症(idiopathic cytopenia of undetermined significance,ICUS)。一些ICUS患者 可逐渐发展为典型MDS,因此应严密监测,随访过程中如患者出现典型的细胞遗传学异常,即使仍然缺乏原始细胞增加及细胞发育异常的表现,应诊断为MDS。MDS诊断依赖于多种实验室检测技术的综合使用,其中骨髓细胞形态学和细胞遗传学检测技术是MDS诊断的核心。MDS的主要诊断方法见表16-3-23。
表16-3-22 MDS维也纳最低诊断标准

表20-3-23 MDS的主要诊断方法

注:AML:急性髓系白血病;PNH:阵发性睡眠性血红蛋白尿症;LGL:大颗粒淋巴细胞白血病;SNP-array:单核苷酸多态性微阵列
(二)鉴别诊断
1.慢性再障 再障多为全血细胞减少,部分患者骨髓有局灶性增生,而MDS多为增生性,少数为增生低下,需要鉴别。慢性再障淋巴细胞相对增多,骨髓象中红系、粒系及巨核系形态无异常,且巨核细胞常减少或缺如,骨髓小粒主要是非造血细胞。染色体检查无异常。MDS骨髓一般有红系、粒系及巨核系的增生,并有病态造血,骨髓小粒主要是造血细胞。常有染色体异常。
2.巨幼细胞贫血 MDS会出现巨大红细胞及巨幼样红细胞,应与巨幼细胞贫血鉴别。后者常可找到引起叶酸或(和)维生素B 12 缺乏的原因,血清叶酸或(和)维生素B 12 测定降低,红、粒、巨核细胞均可巨幼变,幼红细胞PAS染色阴性,补充叶酸和(或)维生素B 12 病情可以改善。
3.红白血病(M 6 )MDS骨髓红系比例可明显增加,有时可达≥50%有核细胞,该时需注意和纯红白血病、急性红白血病及AML伴MDS相关改变作鉴别,如外周血或骨髓原始细胞<20%,骨髓非红系细胞中原始细胞<20%应诊断为MDS。

(资源67) “疑难血液病临床和细胞形态学讨论”之七(病例)
其他需要鉴别的因素或疾病还有:
4.接受细胞毒性药物、细胞因子治疗或接触有血液毒性的化学制品或生物制剂等;重金属中毒、过度饮酒。
5.慢性病性贫血(感染、非感染性炎症或肿瘤)、慢性肝病、HIV感染。
6.自身免疫性血细胞减少、甲状腺功能减退或其他甲状腺疾病。
7.其他可累及造血干细胞的疾病,原发性骨髓纤维化(尤其需要与伴有纤维化的 MDS 相鉴别)、大颗粒淋巴细胞白血病(LGL)、阵发性睡眠性血红蛋白尿症(PNH)、急性白血病[尤其是伴有血细胞发育异常的形态学特点的患者或急性髓系白血病(AML)-M 7 ]及其他先天性或遗传性血液病(如先天性红细胞生成异常性贫血、遗传性铁粒幼细胞性贫血、先天性角化不良、范可尼贫血、先天性中性粒细胞减少症和先天性纯红细胞再生障碍性贫血等)。
【治疗】
MDS患者自然病程和预后的差异性很大,应根据病情的不同进行个体化的治疗。低危组MDS患者的治疗目标是改善造血、提高生活质量(图16-3-4),高危组 MDS治疗目标是延缓疾病进展、延长生存期和治愈(图16-3-5)。治疗方法和措施主要有对症支持治疗、激素治疗、细胞因子治疗、诱导分化治疗、去甲基化药物治疗、免疫调节剂治疗、化疗、免疫抑制剂治疗、造血干细胞移植等。可酌情选用下列方法。
(一)对症支持治疗
对一些病情较稳定低危患者,以支持疗法为主,同时密切观察病情及血常规的变化。对大多数有较严重贫血(Hb<60g/L)的患者,可以输红细胞以改善贫血。部分RAS患者使用维生素B 6 (100~200mg/d),对RA-S患者可能有一定疗效。对于红细胞输注依赖的患者应定期监测血清铁蛋白(SF)水平、累及输血量和器官功能(心、肝、胰腺),评价铁超负荷程度。铁过载(SF>1000μg/L)需要祛铁治疗,最常用的祛铁剂有:祛铁胺(desferrioxamine),20~60mg/kg,每次静脉或输液泵持续皮下输注8~24小时,每周连续应用5~7天,至SF<1000μg/L。口服祛铁剂地拉罗司分散片(Deferasirox),剂量为20~40mg/(kg·d),每天相同时间顿服,充分溶解于白水或纯果汁(不含碳酸)后口服,注意该药不可溶解于牛奶或碳酸类饮料中,肌酐清除率<40ml/min的患者禁用。对严重血小板减少(<10×10 9 /L)伴有明显出血倾向时输血小板悬液。有感染时应给予抗生素治疗,控制感染。
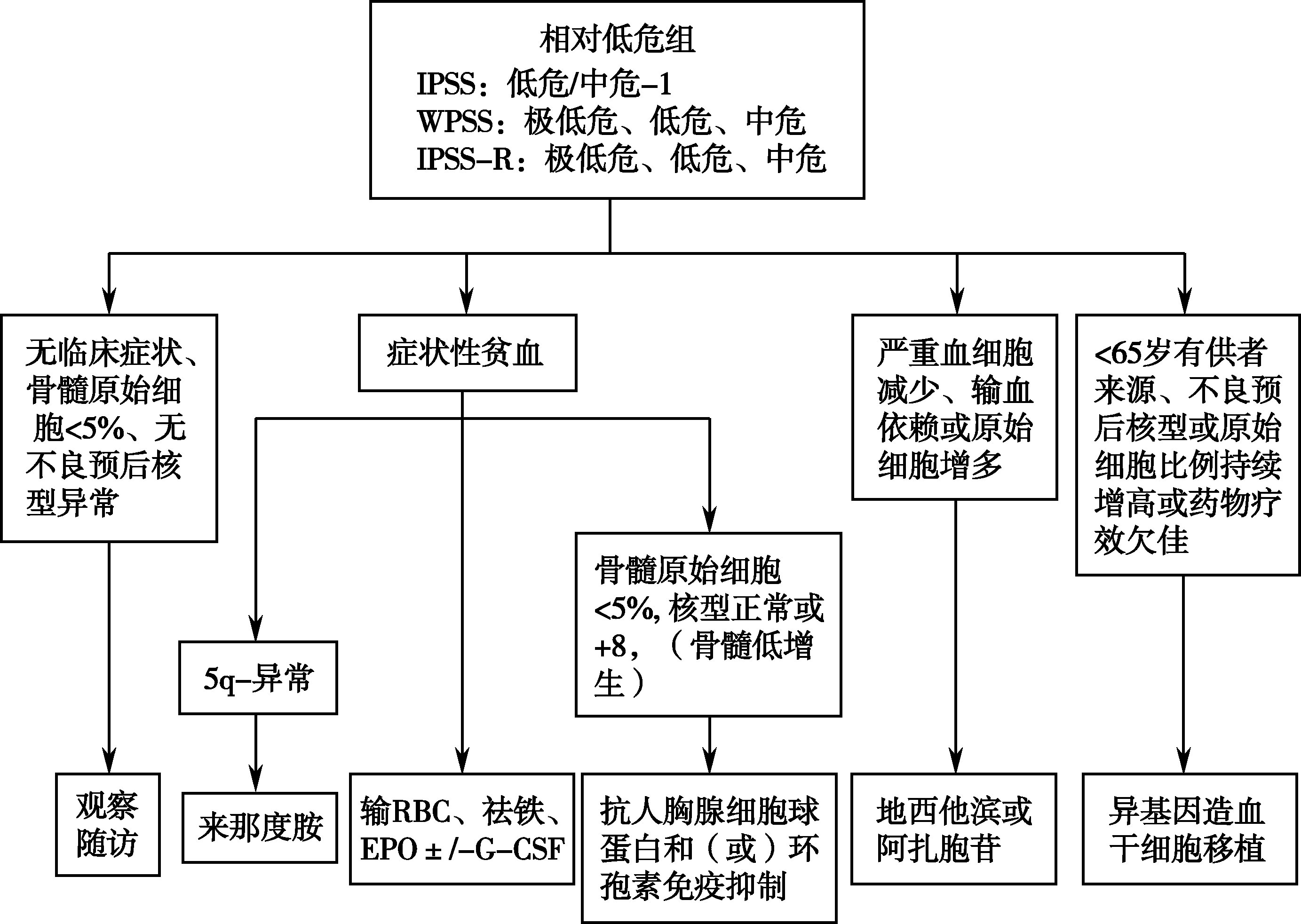
图16-3-4 相对低危组骨髓增生异常综合征患者的治疗
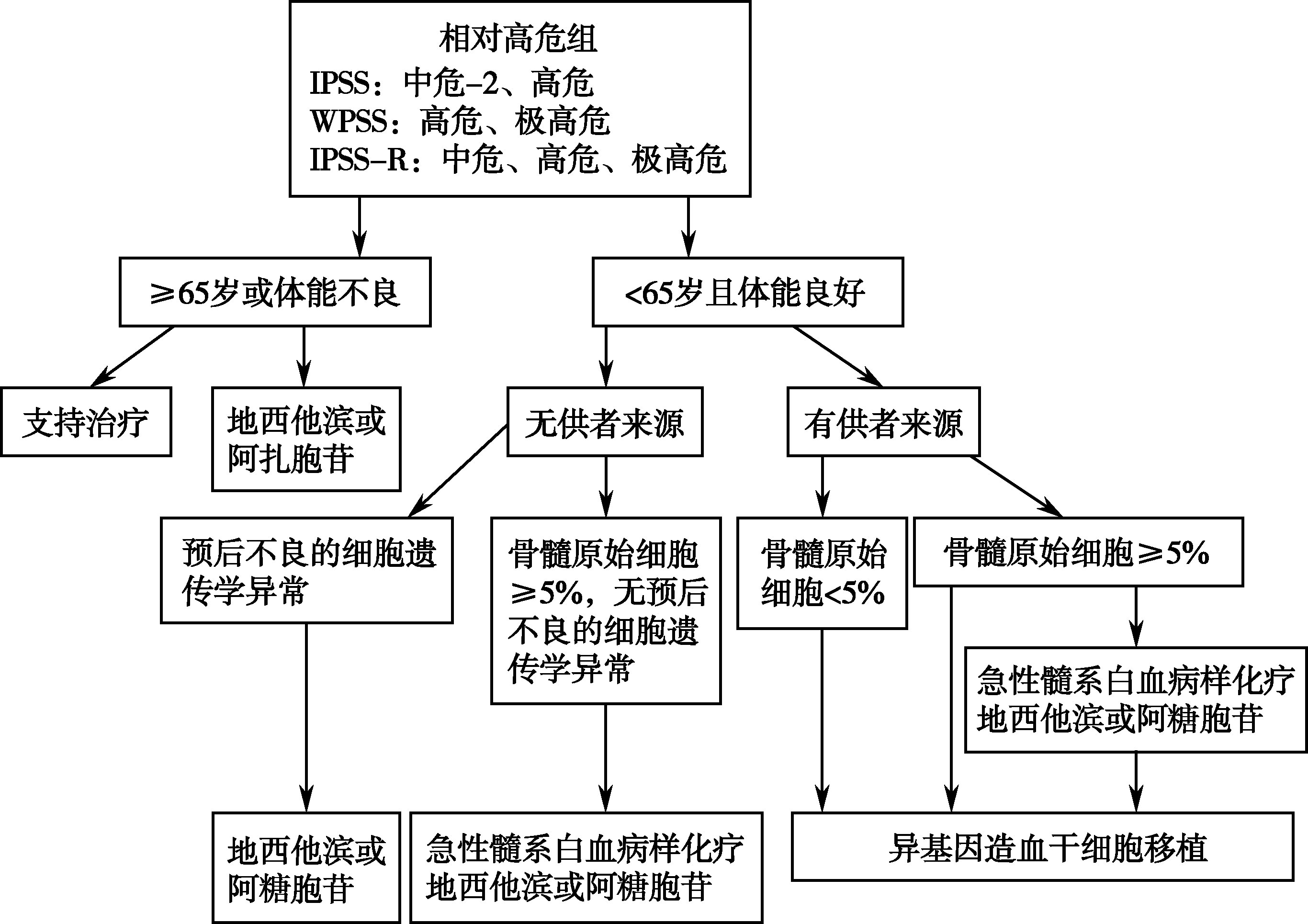
图16-3-5 相对高危组骨髓增生异常综合征患者的治疗
(二)激素治疗
适用于伴有血细胞减少的RA、RARS及原始细胞比例低的RAEB型,对一部分患者可能有效。
1.雄激素
参见第二章第二节“再生障碍性贫血和其他骨髓衰竭综合征”。
2.糖皮质激素
泼尼松:剂量lmg/(kg·d),疗程3个月以上,有效率低于10%。亦有主张甲泼尼龙冲击疗法剂量1g/d,连用3天。对少数患者有效。
(三)细胞因子
细胞因子可以刺激骨髓中残存的正常祖细胞的增殖分化,和(或)诱导 MDS克隆转化为正常造血细胞,促进强化疗后患者造血功能的恢复,减少因骨髓抑制导致感染和出血的危险。
1.促红细胞生成素(EPO)
剂量50~300U/(kg·d),皮下注射,隔日1次或每周3次,疗程3~12个月。对于轻度贫血的RA、RARS患者且血清EPO水平较低,体外造血细胞培养CFU-E,BFU-E对EPO有反应者效果较好,血清EPO水平>500U/L者疗效较差。
2.粒-巨噬系和粒系集落刺激因子(GM-CSF,G-CSF)
不推荐长期单独使用,仅在反复或伴有耐药感染的粒细胞缺乏患者中可以使用剂量60~200μg/(m 2 ·d),皮下注射,疗程视病情需要确定,一般2~8周。对于原始细胞过多的RAEB患者应在化疗的基础上使用。
3.白细胞介素-3(IL-3)
剂量50~200μg/(m 2 ·d),皮下注射,疗程2~8周。
4.促血小板生成素(TPO)
剂量1μg/(kg·d),皮下注射,主要用于血小板有明显减少者,7~14天。
5.白细胞介素-11(IL-11)
用于血小板减少的治疗,25~50μg/(kg·d),皮下注射,7~14天。
(四)诱导分化剂
其作用机制为刺激 MDS异常造血克隆转变为正常克隆及促进来源于的异常克隆的各阶段幼稚细胞进一步分化为成熟细胞。适用于各型MDS患者。
1.全反式维甲酸
为维生素A的衍生物,可促进早期粒细胞的分化,抑制白血病细胞的增殖。剂量30~40mg/d,疗程一般1~3个月。亦有人主张小剂量长疗程,10~20mg/d,3~6个月,效果可能更好。不良反应:皮肤过度角化,口唇干裂,头疼,关节肌肉酸痛,肝功能损害等。
2.1,25(OH) 2 D 3
可抑制白血病细胞增殖和促进分化。剂量2.5~15.0μg/d,疗程2~6个月,少数人有效。不良反应:高血钙,常限制其长期或大剂量服药。其他的不良反应包括厌食、恶心、烦渴多尿、嗜睡,停药后可恢复。
(五)去甲基化药物
5-氮杂胞苷(5 AC)和地西他滨(DAC)为DNA甲基转移酶抑制剂,可逆转DNA过度甲基化使因过度甲基化而致缄默的基因重新表达。5AC 75mg/(m 2 ·d)皮下注射,连用7天,4周为1疗程,4~6个疗程,有效者可继续使用。DAC 20mg/(m 2 ·d),连用5天,4周为1疗程,4~6疗程,有效者可继续使用;DAC亦可和其他化疗药联合使用。
(六)免疫调节剂治疗
沙利度胺(反应停):50~200mg,每晚1次。来那度胺,其抗肿瘤和免疫调节作用比沙利度胺更强,而不良反应明显低于沙利度胺,推荐剂量为10mg/d,连用21天,4周一疗程。对于伴有5q-的IPSS-低危或中危1组MDS患者,如存在输血依赖性贫血、且对细胞因子治疗效果不佳,可应用来那度胺治疗。伴有5q-的MDS患者,若出现下列情况不建议使用来那度胺:骨髓原始细胞比例>5%;复杂染色体异常;IPSS-中危2 或高危组;检出 p53 基因突变。
(七)化疗
1.小剂量化疗
常采用小剂量阿糖胞苷(Ara-C)10~20mg/(m 2 ·d),14~21天一疗程;三尖杉碱(H)1mg/d,10~14天一疗程;阿克拉霉素3~14mg/(m 2 ·d),7~10天一疗程;或鬼臼乙叉苷(VP16),25~35mg/(m 2 ·d),7~10天一疗程。预激方案为在小剂量阿糖胞苷(10mg/m 2 ,每12小时1次,皮下注射,×14天)基础上加用G-CSF,并联合阿克拉霉素或高三尖杉酯碱或去甲氧柔红霉素,常用于老年或身体功能较差的相对高危组患者。
2.联合化疗
按照AML标准方案治疗,具体可参见第三章第二节“急性白血病”章节的相关治疗部分。
(八)免疫抑制剂治疗
抗胸腺淋巴细胞球蛋白(ATG)与环孢素,通过抑制CD8细胞来调节MDS的免疫反应。ATG(马抗)剂量15~40mg/(kg·d),连用4~5天;环孢素剂量3~5mg/(kg·d),3~6个月。ATG和环孢素可以合用。≤60岁的IPSS低危或中危-1、骨髓原始细胞比例<5%或骨髓增生低下、正常核型或单纯+8、存在输血依赖、HLA-DR15 + 或存在PNH克隆的患者适用。原始细胞>5%、-7或有复杂染色体等情况下不宜使用。
(九)其他药物
1.砷剂
三氧化二砷对部分患者有效。剂量10mg/d,静脉滴注,疗程4~6周。不良反应主要为肝功能损害。
2.丙戊酸钠
具有组蛋白去乙酰化酶抑制剂活性,5~10mg/(kg·d)口服。
3.氨磷汀(阿米福汀,Amifostine)
一种磷酸化的有机硫醇,其体内代谢产物有抗氧化保护细胞作用,对骨髓造血前体细胞亦有促进生长作用。剂量200~400mg/m 2 ,每周3次,3~4周一疗程。主要不良反应为恶心、呕吐。
(十)造血干细胞移植
异基因骨髓移植(allo-HSCT)是目前可能治愈本病的一种治疗方法,其适应证:①年龄<65岁、相对高危组MDS患者;②年龄<65岁、伴有严重血细胞减少、经其他治疗无效的中低危患者。近年,减低强度的预处理方案受到重视,该法降低移植相关死亡率,而疗效基本不受影响。拟行allo-HSCT的患者,如骨髓原始细胞≥5%,在等待移植的过程中可应用化疗或联合去甲基化药物桥接allo-HSCT。
MDS国际工作组(International Working Group,IWG)于2000 年提出国际统一疗效标准,2006 年又进一步修订,使不同临床治疗方案结果间具有可比性。MDS的治疗反应包括以下四种类型:改变疾病的自然病程、细胞遗传学反应、血液学改善和改善生存质量(表16-3-24)。
表16-3-24 MDS国际工作组(IWG)疗效标准

续表

注:ANC:中性粒细胞绝对计数
【病程与预后】
由于MDS亚型的异质性及其临床表现的多样性,MDS患者的生存率变化也非常大,影响预后的因素包括年龄、疾病的自然病程、临床特征、骨髓及外周血原始细胞的水平、细胞遗传学异常以及细胞减少的系列数等。此外,病理特征如骨髓中Auer小体的存在,ALIP和假Pelger-Hüet畸形等对预后也产生影响。1997年,英美法德西班牙等国的血液工作者提出了MDS患者预后的国际积分系统,已广泛用于临床(表16-3-25)。这一系统把患者分为相对低危(低危0分;中危-1,0.5~1分)和相对高危(中危-2,1.5~2分;高危,≥2.5分)预后组(表16-3-26)。在中位生存期和AML转化危险性确定后,发现年龄与生存也相关,60岁与>60岁年龄组预后不同(表16-3-27)。2012年,MDS预后国际工作组依据5个MDS数据库,共7012例MDS患者的研究结果,对IPSS预后评分系统进行了修订,对染色体核型、骨髓原始细胞数和血细胞减少程度进行了细化分组积分(表16-3-28)。核型分析结果是IPSS-R分类最重要的参数,共分为五个级别:极低危:≤1.5分;低危:>1.5~3分;中危:>3~4.5分;高危:>4.5~6分;极高危:>6分。
表16-3-25 MDS的国际预后积分系统(IPSS)
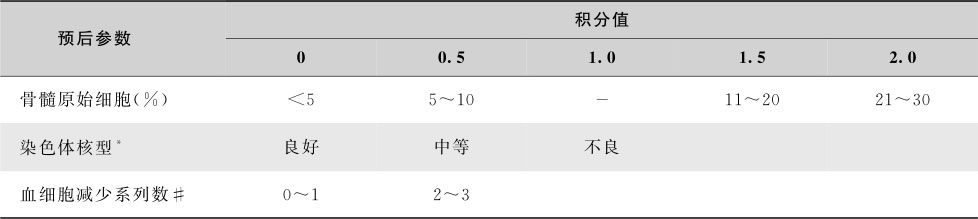
注: * 良好:正常核型,-Y,del(5q),del(20);不良:复杂核型(≥3种异常核型改变)或7号染色体异常;中等:其他异常。 # 血红蛋白<100g/L、中性粒细胞<1.5×10 9 /L、血小板<100×10 9 /L。
表16-3-26 按IPSS分组的MDS患者的中位生存和AML转化时间
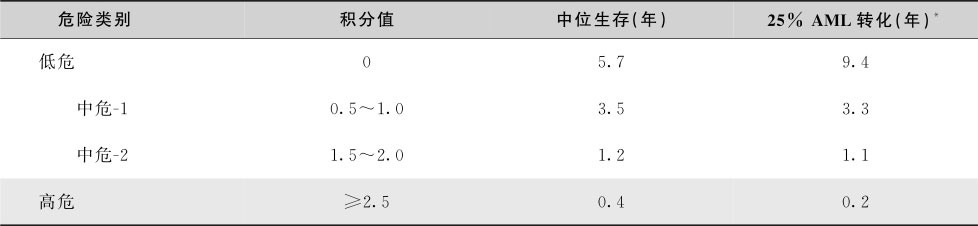
注: * 直至该组中25%的患者转化为AML的时间
表16-3-27 按IPSS及年龄分组的MDS患者的中位生存和25%AML转化时间
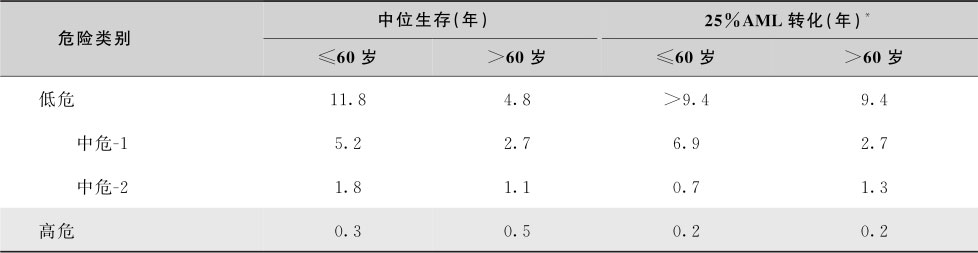
注: * 直至该组中25%的患者转化为AML的时间
表16-3-28 MDS的国际预后积分系统(IPSS-R)
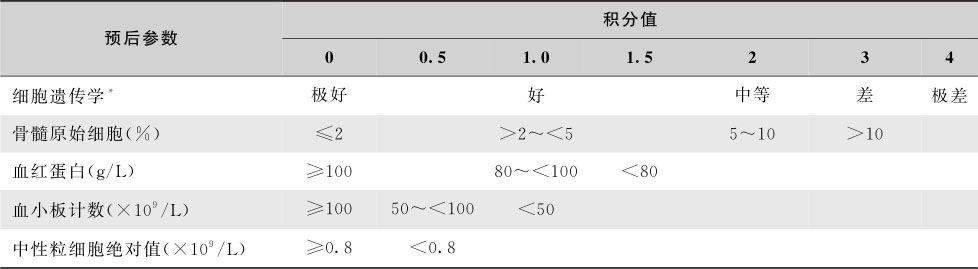
注:* 极好:-Y,11q-;好:正常核型,5q-,12p-,20q-,5q-附加另一种异常;中等:7q-,+8,+19,i(17q),其他1个或2个独立克隆的染色体异常;差:-7,inv(3)/t(3q)/del(3q),-7/7q-附加另一种异常,复杂异常(3个);极差:复杂异常(>3个)
据WHO分型、输血是否依赖、染色体核型情况的WHO预后分析系统(WPSS)把患者分为5组,极低危组(0分)、低危组(1分)、中危组(2分)、高危组(3~4分)、极高危组(5~6分)。2011年修订的WPSS预后评分系统将评分依据中的红细胞输注依赖改为血红蛋白水平(表16-3-29)。
表16-3-29 MDS的WHO预后分析系统(WPSS)
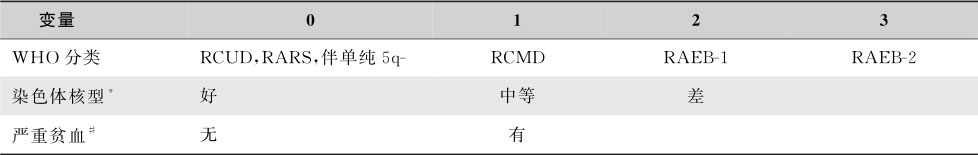
注: * 好:正常核型,-Y,5q-,20q-;差:复杂核型,7号染色体异常;中等:其他异常。 # 男性患者 Hb<90g/L,女性患者Hb<80g/L
主要参考文献
1.张之南,沈悌.血液病诊断及疗效标准.第3版.北京:科学出版社,2007,157-163.
2.Aoki E,Uchida T,Ohashi H,et al.Methylation status of p15INK4B gene in hematopoietic progenitors and peripheral blood cells in myelodysplastic syndromes.Leukemia,2000,14(4):586-593.
3.Nilsson L,Eden P,Olsson E,et al.The molecular signature of MDS stem cells supports a stem origin of 5q myelodysplastic syndromes.Blood,2007,110(8):3005-3014.
4.Greenberg P,Cox C,Le Beau MM,et al.International scoring systems for evaluating prognosis in myelodysplastic syndromes.Blood,1997,89(6):2079-2088.
5.List A,Kurtin S,Roe DJ,et al.Efficacy of lenalidomide in myelodysplastic syndromes.N Engl J Med,2005,352(6):549-557.
6.Valent P,Horny HP,Bennett JM.Definitions and standards in the diagnosis and treatment of the myelodysplastic syndromes:Consensus statements and report from a working conference.Leuk Res,2007,31(6):727-736.
7.Malcovati L,Germing U,Kuendgen A,et al.Time-dependent prognostic scoring system for predicting survival and leukemic evolution in myelodysplastic syndromes.J Clin Oncol,2007,25(23):3503-3510.
8.Brunning RD,Orazi A,Germing U,et al.Myelodysplastic syndromes/neoplasms.in:Swerdlow SH,Campo E,Harris NL,et al,eds.World Health Organization Classification of Tumours.Pathology&Genetics.Tumours of Haematopoietic and Lymphoid Tissues.4th ed.Lyon:IARC Press,2008,88-107.
9.中华医学会血液学分会.骨髓增生异常综合征诊断与治疗中国专家共识(2014年版).中华血液学杂志,2014,35(11):1042-1048.
第六节
骨髓增生异常/骨髓增殖性肿瘤
许小平
骨髓增生异常/骨髓增殖性肿瘤(myelodysplastic/myeloproliferative neoplasm,MDS/MPNs)包括4种独立的疾病:①慢性粒-单核细胞白血病;②不典型慢性髓性白血病;③幼年型慢性粒-单核细胞白血病;④不能分类的MDS/MPN。此外,对于诊断符合 MDS-RARS标准,但同时伴有血小板显著增多(血小板计数>450×10 9 /L)者,也将其列为一种暂定的疾病实体,命名为血小板显著增多的伴环状铁粒幼细胞难治性贫血(refractory anemia with ring sideroblasts associated with marked thrombocytosis,RARS-T)。MDS/MPNs患者造血系统的共同特征是既可有血细胞减少和发育异常的MDS表现,同时也可有髓系其他系列血细胞不同程度增生并伴发髓外造血的MPN特点。
一、慢性粒-单核细胞白血病
(一)临床表现
在 WHO(2001)髓系肿瘤分类提出之前,FAB分型将慢性粒-单核细胞白血病(chronic myelomonocytic leukemia,CMML)列为 MDS的一个亚型。CMML约占MDS的31%左右,中位年龄为65~75岁,男女比例为1.5~3.0∶1。常见的症状为虚弱、体重减轻、发热、盗汗,感染、出血。多数患者诊断时外周血白细胞计数增多,部分患者也可正常或轻度下降,并伴有不同程度的中性粒细胞减少。单核细胞绝对值>1×10 9 /L,常持续3个月以上。肝、脾增大在白细胞增多的患者中可高达50%。
(二)实验室检查
外周血单核细胞增多为CMML的特征性改变,单核细胞计数>1×10 9 /L,通常为(2~5)×10 9 /L,也可>80×10 9 /L,单核细胞百分比几乎总>10%。单核细胞一般为成熟细胞,形态不典型,可表现为异常颗粒、核分叶或细而稀疏染色质。可见原始及幼稚单核细胞。白细胞可增高、正常或轻度减低。早幼粒、中幼粒细胞常<10%。有时可有轻度嗜碱性粒细胞和嗜酸性粒细胞增多。大部分患者可有粒细胞发育异常,轻度正细胞性或大细胞性贫血,常有中度血小板减少。
外周血与骨髓细胞免疫表型检测通常表达粒单核细胞系抗原CD33及CD14、CD68、CD64。CD34 + 细胞比例增多提示向急性白血病早期转化。
20%~40%的患者有克隆性细胞遗传学异常。最常见的染色体异常包括+8,-7/del(7q)及12q结构异常,11q23异常并不常见。
(三)诊断
WHO(2008)分类提出的CMML诊断标准如下:
1.外周血单核细胞持续性>1×10 9 /L。
2.无Ph染色体或 BCR-ABL1 融合基因。
3.无 PDGFRA 或 PDGFRB 重排(特别应除外嗜酸性粒细胞增多的病例)。
4.外周血和骨髓原始细胞<20%。
5.一系或一系以上髓系发育异常。
如髓系发育异常缺乏或轻微,但其他条件符合,并且:①骨髓细胞有获得性、克隆性染色体异常;②单核细胞持续增多3个月以上;③排除其他所有引起单核细胞增多的原因。仍可诊断CMML。
依据外周血及骨髓原始细胞数不同,CMML可分为两型:
CMML-1:原始细胞(包括幼稚单核细胞)外周血<5%,骨髓<10%。
CMML-2:原始细胞(包括幼稚单核细胞)外周血5%~19%或骨髓10%~19%;或外周血或骨髓原始细胞+幼稚单核细胞数虽未达诊断标准,但可找到Auer小体。
此外,MDS/MPN 伴有与t(5;12)(q31~33;p12)及 ETV 6 -PDGFRB 融合基因相关的嗜酸性粒细胞增多的患者,以前也诊断为CMML,目前则认为是另一种独立的疾病实体。
(四)治疗与预后
小剂量化疗和大剂量强烈化疗效果均不令人满意。异基因造血干细胞移植是唯一有希望治愈本病的手段,对于年龄较轻者应首先考虑异基因造血干细胞移植。文献报道10年的无进展存活(PFS)为38%,复发率为27%。但由于慢性粒-单核细胞白血病患者多为老年人,一般不能耐受强烈的预处理方案,临床上多以支持治疗和对症处理为主。羟基脲一般用于脾脏增大伴白细胞计数升高患者;去甲基化治疗的整体反应率(ORR)为40%;托扑替康为基础的方案或小剂量阿糖胞苷完全缓解率为27%~40%。以上治疗存活时间大多不超过2年。CMML只有极少数患者呈惰性病程,多数进展迅速,大约15%~30%病例转为急性白血病。外周血与骨髓原始细胞百分率是决定生存的最重要因素。
二、不典型慢性粒细胞白血病
不典型慢性粒细胞白血病(atypical chronic myeloid leukemia,aCML)是一种同时具有 MDS和 MPN特征的慢性粒细胞白血病,其特点是具有发育异常的幼稚和成熟中性粒细胞增多,但白血病细胞无Ph染色体或 BCR-ABL1 融合基因。
(一)临床表现
本病罕见。患者多为老年人,诊断时中位年龄>60岁。男女比例约为1∶1。因白细胞计数增高、外周血出现中性粒细胞前体细胞就诊者很常见,脾脏增大见于约70%的病例。患者常有贫血或血小板减少。
(二)实验室检查
患者外周血白细胞数≥1.3×10 9 /L,中位数为(24~96)×10 9 /L,有的患者白细胞计数可>300×10 9 /L。外周血原始细胞通常<5%,不超过20%。幼稚粒细胞通常为10%~20%或更多,尽管单核细胞绝对值可增多,但其百分率很少超过10%,嗜碱性粒细胞增多常不明显。中度贫血和血小板减少多见。
骨髓增生极度活跃,以中性粒细胞及其幼稚粒细胞增生为主,原始细胞可中度增多,但<20%,骨髓中粒系形态改变与外周血中相似,巨核细胞可减少、正常或增多,大部分患者有巨核细胞发育异常。红系增生程度不定,通常粒红比例>10∶1,但有些病例幼红细胞可>30%,至少50%的患者存在红系发育异常。部分患者初诊时或疾病晚期网状纤维增多。
80%患者有细胞遗传学异常,但未发现有特征性的细胞分子遗传学改变。最常见的为+8,del(20q),无Ph染色体或 BCR-ABL1 融合基因。
(三)诊断标准
WHO(2008)分类诊断标准:
1.因成熟和幼稚中性粒细胞增多引起的外周血白细胞增多(WBC≥13×10 9 /L)。
2.粒细胞具有显著发育异常。
3.无Ph染色体或 BCR-ABL1 融合基因,无 PDGFRA 或 PDGFRB 重排。
4.外周血中性粒细胞前体细胞(早幼粒、中幼粒、晚幼粒细胞)≥10%。
5.轻微嗜碱性粒细胞绝对数增多,嗜碱性粒细胞<2%。
6.无或轻微单核细胞绝对数增多;单核细胞<10%。
7.骨髓增生极度活跃,粒细胞增殖及粒细胞发育异常,伴有或不伴有红系和巨核系发育异常。
8.外周血或骨髓原始细胞<20%。
(四)治疗与预后
羟基脲对于缓解白细胞增高、脾脏增大引起的症状有效,整体反应率(ORR)在80%左右,但持续有效时间仅能维持2~4个月。α-干扰素300~600万U/d,43%患者有治疗反应,也有少数病例获得完全或部分缓解。最近有采用地西他滨20mg/m 2 ×5天,1个化疗周期获得血液学反应,3个周期原始细胞下降的个案报道。aCML中位生存期14~29个月。年龄>65岁、女性、白细胞计数>50×10 9 /L、血小板减少及Hb<10g/L为预后不良因素。约20%~45%的病例转化为急性白血病。异基因造血干细胞移植可能有利于改善预后。
三、幼年型粒-单核细胞白血病
幼年型粒-单核细胞白血病(juvenile myelomonocytic leukemia,JMML)主要以粒细胞及单核细胞增殖为特征,常有红系及巨核系异常。儿童JMML年发病率约为0.12/10万,占所有儿童白血病不足2%~3%,占所有14岁以下儿童骨髓增生异常及骨髓增殖性疾病的20%~30%。初诊年龄为1个月至早期青春期,75%患者年龄<3岁,中位发病年龄为2岁。男性发病率约为女性的2倍。一些病例常与神经纤维瘤病和Noonan综合征共存,因此推测JMML发病可能与RAS信号通路异常有关。
(一)临床表现
患者临床症状多与单核细胞大量增生并浸润全身器官有关。可见全身不适、发热,肝、脾显著肿大,近半数患者有淋巴结肿大,扁桃体也可因单核细胞浸润而肿大,肺和肠道因细胞浸润也可出现相应症状。血小板常减少,出血较常见。约1/4患者出现皮疹。
(二)实验室检查
外周血可见贫血和血小板减少,白细胞计数增多。白细胞平均值在25×10 9 /L~30×10 9 /L,少数患者可>100×10 9 /L。主要为中性粒细胞和单核细胞增多,原始细胞(包括原始单核细胞与幼稚单核细胞)<20%。少数患者有嗜碱性粒细胞和嗜酸性粒细胞增多。常见有核红细胞。HbF可升高。
骨髓穿刺和活检增生极度活跃,主要以粒系细胞增生为主,但有的患者幼稚红细胞可高达50%左右。单核细胞一般为5%~10%,亦可>30%。原始细胞(包括原始单核细胞与幼稚单核细胞)<20%,无Auer小体。病态造血通常较轻微。
细胞遗传学检查25%的患者有7号染色体单体,其他染色体核型异常为10%,65%患者为正常核型。无Ph染色体或 BCR-ABL1 融合基因。
(三)诊断
WHO(2008)分类提出的JMML的诊断标准:
1.外周血单核细胞增多>1×10 9 /L。
2.外周血和骨髓原始细胞(包括幼稚单核细胞)<20%。
3.无Ph染色体或 BCR-ABL1 融合基因。
4.加下述2项或2项以上;①HbF较同年龄正常人增高;②外周血出现幼稚粒细胞;③白细胞数>10×10 9 /L;④克隆性染色体异常(如7号染色体单体);⑤体外髓系祖细胞对GM-CSF敏感性增高。
(四)治疗与预后
JMML虽然很少转变为急性白血病,但如不治疗,将很快危及生命。有报告不行异基因造血干细胞移植的患者中位生存期为1年左右。诊断时血小板计数低、年龄>2岁,以及高Hb F预示生存期短。异基因造血干细胞移植的5年无事件存活(EFS)为52%。
四、骨髓增生异常/骨髓增殖性肿瘤,不能分类
骨髓增生异常/骨髓增殖性肿瘤,不能分类(myelodysplastic/myeloliferative neoplasm,unclassifiable,MDS/MPN,U)是指患者初次诊断时具有MDS/MPN的临床和实验室检查特征,但不符合上述3类诊断标准者。因此,MDS/MPN,U具有高度异质性。
(一)临床表现
本病约占所有髓系肿瘤的5%以下。中位发病年龄71岁,男性多见,约为女性的2倍左右。部分病例有肝、脾大。实验室检查多数患者有大红细胞性贫血,骨髓呈现一系或多系有效增殖伴发育异常。既可血小板增多(血小板计数>450×10 9 /L),也可白细胞增多(白细胞计数>13×10 9 /L),中性粒细胞发育异常,血小板体积巨大或颗粒过少,外周血和骨髓原始细胞<20%,若>10%提示可能已转化为更具侵袭性的阶段。骨髓活检增生极度活跃,一系或多系髓系细胞增殖,至少同时有一系细胞呈现发育异常。骨髓纤维化也常可见到。大约20%~30%患者 JAK2V617F 阳性。至今尚未发现MDS/MPN,U的标志性细胞遗传学改变,但分子细胞遗传学检查有助于与CML、5q-综合征以及伴 PDGFRA 、 PDGFRB 或 FGFR1 重排的髓系肿瘤进行鉴别。
(二)诊断标准
WHO(2008)提出的诊断标准如下:
有 MDS各型(RCUD、RARS、RCMD、RAEB)之一的临床、实验室及形态学特点,骨髓和外周血原始细胞<20%,同时具备:
1.有明显的骨髓增殖性疾病表现,即血小板数≥450×10 9 /L,伴巨核细胞增多,或白细胞≥13.0×10 9 /L;伴有或不伴有明显的脾大。
2.无先前潜在的MPN或MDS病史,无近期可以导致骨髓增生异常或骨髓增殖表现的细胞毒或细胞因子治疗的病史,无Ph染色体或 BCR-ABL1 融合基因、无 PDGFRA 、 PDGFRB 或 FGFR1 重排,无del(5q),t(3;3)(q21;q26)或inv(3)(q21;q26)。
3.患者有原发性骨髓增殖性疾病和骨髓增生异常的混合表现,并且不能归入任何一种独立类型的MDS、MPN或前述3类MDS/MPN中。
在WHO髓系肿瘤分类中特别强调MDS/MPN,U不能用于过去明确诊断为MPN而向更侵袭性病程转化伴有血细胞发育异常的病例。但MDS/MPN,U实际上可能包括了过去未发现的MPN慢性期的患者初诊时表现为以骨髓增生异常为特征转化期的患者。
对于有MDS-RARS临床和形态学表现的患者如伴有血小板显著增多(血小板计数>450×10 9 /L),称血小板显著增多的伴环状铁粒幼细胞难治性贫血(refractory anemia with ring sideroblasts associated with marked thrombocytosis,RARS-T),因目前尚不清楚RARS-T是否为一种独立的疾病实体,还是两种独立疾病(RARS和ET)共存,WHO将其作为一种暂定的疾病名称单独列出。已发现这组患者多有 JAK2V617F 突变,少数为 MPLW515K / L 突变;SF3B1突变可高达72%。但细胞遗传学检查有孤立性del(5q)、t(3;3)(q21;q26)或inv(3)(q21;q26)的患者不应该诊断RARS-T。此外,初诊时不伴有环状铁粒幼细胞的MPN患者,以后在治疗或疾病进程中出现环形铁粒幼细胞也不应诊断为RARS-T。
(三)治疗与预后
美国MD Anderson癌症中心(MDACC)的经验显示,在非造血干细胞移植治疗的患者中,去甲基化药物(HMA)治疗的整体存活(OS)较α-干扰素、环孢素、沙利度胺、雷那度胺和抗胸腺球蛋白等药物延长(16.4个月 vs .11.5个月)。另有HMA联合JAKs抑制剂ruxolitinib的临床试验也正在进行中。由于RARS-T频发SF3B1突变,抑制剪接体的治疗策略可能会使这部分患者更多获益。
主要参考文献
1.Mughal TI,Cross NCP,Padron E,et al.An International MDS/MPN Working Group's perspective and recommendations on molecular pathogenesis and clinical characterization of myelodysplastic/myeloproliferative neoplasms.Heamatologica,2015,100(9):1117-1130.
2.Savona MR,Malcovati L,Komrokji R,et al.An international consortium proposal of uniform response criteria for myelodysplastic/myeloproliferative neoplasms(MDS/MPN)in adults.Blood,2015,125(12):1857-1865.
3.Swerdlow SH,Campo E,Harris NL et al.(eds).WHO Classification of Tumors of Haematopoietic and Lymphoid Tissues.4th ed.Lyon:IARC,2008,75-86.
第七节
类白血病反应
陈波斌
内、外源性刺激均可导致外周血白细胞反应性增多。白细胞计数大于11.0×10 9 /L时,临床上可诊断为白细胞增多症(leukocytosis)。并非由白血病引起的外周血白细胞显著增多和(或)出现幼稚的血细胞而类似白血病血象者,称为类白血病反应(leukemoid reaction,LR)。类白血病反应是由非白血病原因诱导某些骨髓细胞释放细胞因子,引起的正常骨髓的极度增生反应。随原发病治疗好转,刺激因素消除,类白血病反应也迅速消失。因此,类白血病反应本身并不需要治疗,关键在于治疗原发病。其临床重要性在于不要误诊。临床上以中性粒细胞类白血病反应最常见,由淋巴细胞、单核细胞或嗜酸性粒细胞等形成的较少见。必须注意在小儿,由于骨髓造血功能未成熟,易受各种病理因素刺激,因而较易发生类白血病反应。
一、中性粒细胞类白血病反应
成人周围血中性粒细胞绝对数大于7.5×10 9 /L时,临床上可诊断为中性粒细胞增多症(neutrophilia)。非白血病引起的周围血中性粒细胞极度升高,白细胞计数达50×10 9 /L以上和(或)出现幼稚粒细胞,称之为中性粒细胞类白血病反应(neutrophilic leukemoid reaction,NLR)。
在急性炎症反应时,骨髓中单核巨噬细胞、血管内皮细胞、成纤维细胞和淋巴细胞等分泌细胞因子如G-CSF、IL-3、干细胞因子和GM-CSF等增多,促进髓系祖细胞向中性粒细胞前体细胞的分化和增殖,导致中性粒细胞显著增多,核左移和幼稚粒细胞被释放到外周血液。类白血病反应时中性粒细胞异常增多与骨髓分裂池和贮备池的扩大,边缘池向循环池迁移增多及循环池中性粒细胞向组织迁移减少有关。某些肿瘤细胞也可产生细胞集落刺激因子,刺激造血细胞增生、分化、释放。毒素、缺氧、免疫反应、化学物质等可损伤骨髓毛细血管内皮细胞,使髓血屏障受损,导致部分幼稚细胞进入血液循环。
【病因】
中性粒细胞增多症和中性粒细胞类白血病反应的病因如下:
(一)感染
可见于化脓性球菌感染如金黄色葡萄球菌、溶血性链球菌、肺炎双球菌、淋球菌、脑膜炎双球菌等;部分杆菌感染如大肠埃希菌、假单胞菌、白喉杆菌、土拉杆菌;部分真菌(球孢子菌、放线菌等);钩端螺旋体;以及部分病毒如水痘、脑炎和肺炎、脊髓灰质炎、天花、流行性出血热等;立克次体(斑疹伤寒)及肝吸虫病等感染。结核性脑膜炎、干酪样坏死灶溃破以及胸膜的累及可引起中性粒细胞显著增多。NLR可见于严重的全身感染,例如在骨髓炎时白细胞可高达75×10 9 /L,脾脓肿时可达100×10 9 /L,败血症时可达112×10 9 /L。全身播散并伴干酪样坏死的结核病例,白细胞计数可达200×10 9 /L,并可在外周血中出现幼稚粒细胞。
(二)其他炎症性疾病、创伤和组织坏死
急性肾小球肾炎、血清病、风湿热、血管炎、某些类型过敏反应(Shwartzman和Arthus反应)和Sweet综合征均可引起中性粒细胞增多。手术后中性粒细胞可持续升高12~36小时。创伤、挤压伤、电击、中暑、低温、缺氧等;心肌梗死、肺梗死、肠梗阻、疝嵌顿引起绞窄及重症胰腺炎等均可引起中性粒细胞显著增多。严重烧伤、创伤和电击可引起NLR,甚至可高达50×10 9 /L以上伴核左移,出现中毒性颗粒和Döhle小体。
(三)非血液恶性肿瘤
生长很快的恶性肿瘤常因肿瘤组织坏死、分泌肿瘤坏死因子(TNF-α)或集落刺激因子直接刺激骨髓,导致中性粒细胞增多。乳腺、前列腺、甲状腺、肺、肝脏、肾和胃肠道等部位的恶性肿瘤患者有时白细胞计数可大于50×10 9 /L,出现类白血病反应,此时应注意骨髓受恶性肿瘤细胞浸润的可能。真性红细胞增多症、原发性血小板增多症和骨髓纤维化等常伴中性粒细胞显著增多。
(四)中毒和药物中毒
内源性中毒包括甲状腺危象、痛风急性发作、糖尿病酮症酸中毒、尿毒症、子痫、肝性脑病等;化学物中毒如铅、汞、砷、苯及其衍生物等;一氧化碳中毒及有机磷中毒;药物中毒如氯化钾、毛地黄、樟脑、安替比林、乙酰苯胺、非那西丁、奎尼丁、松节油、吡啶、联苯三酚等;水母、毒蛇或毒蜘蛛咬伤均可引起中性粒细胞增多。注射外源性蛋白质如伤寒疫苗、内毒素等可引起暂时性白细胞减少,随后约1~2小时后可引起中性粒细胞增多。应用肾上腺皮质激素、锂盐、肾上腺素及集落刺激因子(G-CSF,GM-CSF)等药物均可引起中性粒细胞增多。使用G-CSF或GM-CSF的患者血常规白细胞计数可达30×10 9 /L以上,并出现幼稚粒细胞,类白血病反应在停药后消失。
(五)急性出血、急性溶血和中性粒细胞缺乏恢复期
急性失血后1~2小时就可引起中性粒细胞增多,尤见于出血进入浆膜腔者,如异位妊娠破裂出血、颅脑外伤出血及脾破裂出血可致明显中性粒细胞增多。急性大量出血或严重溶血可导致中性粒细胞的增多甚至可达类白血病的程度。粒细胞缺乏症恢复期血常规检查可出现幼稚粒细胞,骨髓中原始及早幼粒细胞的比例甚至可高达诊断白血病的标准,白细胞计数也可高达30×10 9 /L以上,但此种类白血病血常规及骨髓象仅持续1~3周。
(六)其他
中性粒细胞增多还见于脾切除后、阵发性心动过速、分娩、麻醉、Down综合征、遗传性及特发性中性粒细胞增多症。剧烈运动和强烈情绪激动可引起暂时性中性粒细胞增多。
【实验室检查】
1.血常规
白细胞计数一般在(50~100)×10 9 /L,罕有超过200×10 9 /L者,但结核病引起LR患者有白细胞高达220×10 9 /L的报道,中性粒细胞核象有明显左移。一般幼稚细胞比例不太高,原始细胞少见,常无贫血及血小板减少。恶性肿瘤和结核病引起的LR可有贫血。如出现较多幼稚细胞,也以晚幼粒和中幼粒细胞为主,该时白细胞计数<50×10 9 /L也能诊断LR,血常规酷似慢性粒细胞白血病。少数LR病例白细胞计数可不增高甚至降低,可出现原始及早幼粒细胞,血常规酷似急性白血病,主要见于严重结核病。细胞形态常显示中毒性颗粒,胞质空泡等中毒性改变及Döhle小体,而没有白血病细胞的异形或畸形变化。
2.骨髓象
增生活跃或明显活跃,核象左移,原始及早幼粒细胞并无明显增多(但严重结核病引起LR可有明显增多),无白血病“裂孔”现象,也无多系血细胞病态造血表现,中性粒细胞碱性磷酸酶活性升高。
3.特殊检查
细胞遗传学和分子生物学检查无Ph染色体,无 BCR-ABL 融合基因,血清G-CSF浓度升高,克隆性检测示多克隆造血。
【诊断与鉴别诊断】
诊断NLR的要点:①有明确病因,如严重感染、外伤、出血、溶血、恶性肿瘤及药物史等,并有相应的临床表现;②中性粒细胞计数明显增高,一般要>50×10 9 /L,中性粒细胞核象左移,细胞形态变化,除中毒改变外无细胞畸形和病态造血表现,一般无贫血及血小板减少;③中性粒细胞碱性磷酸酶(NAP)积分增高;④通过细胞遗传学和分子生物学检查能除外慢性粒细胞白血病(CML)和慢性中性粒细胞白血病(CNL);⑤原发病经治疗去除后,血常规随之恢复正常。
中性粒细胞类白血病反应主要应和CML和CNL相鉴别(表16-3-30)。
表16-3-30 中性粒细胞类白血病反应与CML、CNL的鉴别诊断
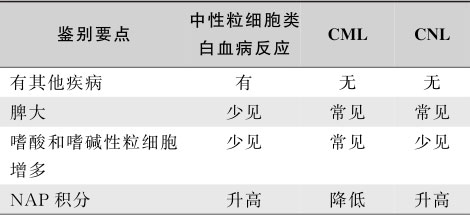
续表
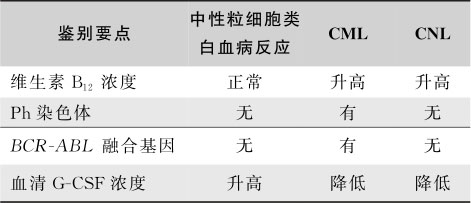
二、单核细胞类白血病反应
外周血单核细胞>0.8×10 9 /L(儿童)及>0.5×10 9 /L(成人),称单核细胞增多症(monocytosis)。单核细胞传递抗原给淋巴细胞,介导细胞毒反应。最有力的促单核巨噬细胞生长的因子是由骨髓基质细胞分泌的M-CSF。单核巨噬细胞向病灶移动的速度缓慢,但和中性粒细胞一样可以有效杀灭细菌,主要杀灭细胞内的病原体,如真菌、病毒等。此外,单核巨噬细胞参与所有类型的肉芽肿性炎症。因此,单核细胞增多可见于结核、梅毒、真菌感染、布鲁菌病、感染性心内膜炎、伤寒和副伤寒、原虫和某些病毒感染(如水痘、登革热)等。也可见于急性感染恢复期、肉芽肿性疾病、结节病以及溃疡性结肠炎、肝硬化及药物反应、霍奇金淋巴瘤和各种肿瘤疾病,单核细胞显著增多最常见于造血系统肿瘤,包括急性和慢性粒单核细胞白血病(CMML)、急性单核细胞白血病和幼年型粒-单核细胞白血病。当白细胞计数大于30×10 9 /L,单核细胞>30%,绝对值超过1×10 9 /L,可有幼稚单核细胞出现,即可诊断单核细胞型类白血病反应,单核细胞型LR多见于结核、感染性心内膜炎、菌痢、传染性单核细胞增多症等,曾有报道菌痢患者白细胞总数达33×10 9 /L,单核细胞占44%。单核细胞型LR需要与CMML进行鉴别,后者可表现为:①骨髓有明显髓系单系或多系病态造血;②肝脾大,脏器存在白血病细胞浸润现象;③骨髓象单核系细胞形态有异形,并经细胞组织化学染色和免疫表型(CD14、CD64、CD68)确认;④Gomori染色显示网状纤维增多,少数合并骨髓纤维化。单核细胞型LR,除单核细胞数量增多外,形态基本正常,为成熟型单核细胞,无异形,其他髓系细胞无病态造血,一般无肝脾大。
三、淋巴细胞类白血病反应
当淋巴细胞计数超过5.0×10 9 /L可诊断为淋巴细胞增多症(lymphocytosis)。轻到中度淋巴细胞增多(淋巴细胞计数<15×10 9 /L)最常见于病毒感染,如传染性单核细胞增多症、病毒性肝炎、传染性淋巴细胞增多症、麻疹、水痘、流行性腮腺炎、巨细胞病毒、腺病毒等,其他的感染如百日咳、弓形虫病、布鲁菌病、结核、伤寒、梅毒也可引起淋巴细胞增多。其中有显著淋巴细胞增多(≥15×10 9 /L)主要见于传染性单核细胞增多症、传染性淋巴细胞增多症及百日咳。急性淋巴细胞白血病和慢性淋巴细胞增殖性疾病亦有显著淋巴细胞增多。此外甲状腺功能亢进、干燥综合征和药物反应(如四环素)也可引起淋巴细胞增多。传染性单核细胞增多症常有显著异形淋巴细胞增多,可超过总淋巴细胞的比例的20%,B淋巴细胞是EB病毒感染的靶细胞,异形淋巴细胞大多数为T淋巴细胞。当白细胞总数显著增多,分类中成熟淋巴细胞占40%以上,绝对值>5.0×10 9 /L,并伴有幼稚淋巴细胞出现时,可诊断为淋巴细胞类白血病反应。据文献报道百日咳引起淋巴细胞绝对值增多,可达(15~25)×10 9 /L,偶有150×10 9 /L,传染性淋巴细胞增多症的严重型淋巴细胞绝对值可达(30~100)×10 9 /L。淋巴细胞型LR有三种类型:①成熟淋巴细胞型,如呈惰性经过,要和慢性淋巴细胞白血病鉴别;②异常淋巴细胞幼稚型,见于传染性单核细胞增多症,要和急性淋巴细胞白血病鉴别,可参见本篇第三章第二节。
四、嗜酸性粒细胞类白血病反应
嗜酸性粒细胞的产生主要受IL-5调节。嗜酸性粒细胞不仅具有吞噬细胞的功能,还能调节超敏反应。当外周血嗜酸性粒细胞>0.5×10 9 /L,诊断为嗜酸性粒细胞增多症(eosinophilia)。临床上有很多原因可引起嗜酸性粒细胞增多(详见本篇第四章第五节“嗜酸性粒细胞增多综合征”),最主要引起反应性嗜酸性粒细胞增多症的病因是寄生虫感染、变态反应性疾病、部分结缔组织病和恶性肿瘤等。外周血嗜酸性粒细胞≥1.5×10 9 /L,并出现幼稚型可诊断为嗜酸性粒细胞类白血病反应。嗜酸性粒细胞型LR主要见于反应性嗜酸性粒细胞增多症,应和克隆性嗜酸性粒细胞增多症相鉴别,后者系指恶性克隆性疾病或骨髓增殖性肿瘤合并嗜酸性粒细胞增多,特别要和慢性嗜酸性粒细胞白血病相鉴别。嗜酸性粒细胞型LR一般外周血无幼稚细胞,骨髓中原始细胞无增多,无嗜酸性粒细胞形态异常,亦无Ph染色体和 PDGFRA 、 PDGFRB 及 FGFR1 重排。
主要参考文献
1.张之南,郝玉书,赵永强,等.血液学.第2版.北京:人民卫生出版社,2011,676-678.
2.Goldman L,Schafer AI.Goldman's Cecil Medicine.25th ed.Philadelphia:Elsevier Saunders,2016,1129-1138.
3.Kaushansky K,Beutler E,Seligsohn U,et al.Williams Hematology.8th ed.New York:McGraw Hill Inc,2010,829-834.

