第四章
骨髓增殖性肿瘤
庄静丽 王宝珍
第一节
骨髓增殖性肿瘤概述

骨髓增殖性肿瘤(myeloproliferative neoplasm,MPN)是2008年WHO新分类定名。系一组克隆性造血干细胞疾病,表现为髓系(粒、红、巨核或肥大)细胞一系或多系增殖,有效性造血,出现外周血细胞增多,肝、脾大,进展缓慢,但可发生急变、骨髓纤维化及无效造血,最后转变为急性白血病。2016年WHO髓系肿瘤分类中,把MPN分为两大类,即慢性性粒细胞白血病 BCR-ABL1 阳性(CML)及 BCR-ABL1 阴性MPN,后者包括慢性中性粒细胞白血病, CSF3R 阳性(CNL),真性红细胞增多症(PV),原发性骨髓纤维化(PMF),原发性血小板增多症(ET),慢性嗜酸性粒细胞白血病,非特定型(CEL,NOS),肥大细胞增生症(通常 KIT 突变)及骨髓增殖性肿瘤,不能分类(MPN,U)。本篇按传统概念将CML和CNL列入白血病章节叙述,其他疾病均在本章叙述。大多数(不是全部)MPN具有编码胞质或受体酪氨酸激酶的基因异常,包括易位或点突变,导致异常酪氨酸激酶,激活信号传导通路,使血细胞异常增殖。例如CML的 BCR-ABL1 融合基因及位于9号染色体( 9P24 ) JAK2 基因突变,后者见于 BCR-ABL1 阴性的MPN,最常见的突变为 JAK2V617F (第617位缬氨酸→苯丙氨酸)。 JAK2V617F 突变见于几乎所有的PV及1/2PMF和ET的病例。 MPL 基因突变可见于3%~5%的ET和8%~10%的PMF患者,常见的类型为 W515 L 和 W515 K ,均发生于 MPL 基因的第10号外显子。进几年的研究显示67%~88%的 JAK2 突变阴性ET或PMF患者可检出 CALR 基因9号外显子的缺失突变,且与 JAK2 、 MPL 突变互排,提示 CALR 突变是MPN重要的驱动突变。
第二节
真性红细胞增多症
真性红细胞增多症(polycythemia vera,PV)简称“真红”,是一种克隆性以红细胞异常增生为主要表现的骨髓增殖性肿瘤。所有病例均有 JAK2V617F 或有其他功能相似 JAK2 基因突变。临床特征有皮肤黏膜红紫、脾大和血管及神经系统症状。血液学的特征为红细胞和全血容量绝对增多,血黏滞度增高,常伴有白细胞和血小板增多。据国外报道发病率为1.9~2.6/10万,无明显地区和国家间的差别。中老年发病较多,50~60岁是发病的高峰,也有少数青年和儿童患者。男性略高于女性。依据病程进展,可分三期:①PV前期:仅有轻度红细胞增多;②显性PV:红细胞显著增多;③衰竭期或PV后骨髓纤维化期(血细胞减少,无效造血,骨髓纤维化,髓外造血,脾功能亢进),少数可向MDS和AML发展。
【病因和发病机制】
真性红细胞增多症的红系祖细胞不依赖促红细胞生成素(EPO)产生内源性红系集落(EEC),并对其他多种造血因子敏感,如PV患者瀑式红系集落形成单位(BFU-E)对白介素-3(IL-3)、粒单核细胞集落刺激因子(GM-CSF)、重组人EPO(rEPO)、胰岛素样生长因子(IGF-I)表现出高度敏感性。PV患者巨核细胞集落(CFU-MK)对TPO高度敏感。PV的造血干/祖细胞在没有TPO条件下能够形成CFU-MK。PV患者存在凋亡异常、造血干细胞细胞遗传学异常及受体和信号传导异常。最近研究表明PV、PMF、ET等与 JAK2 基因突变( JAK2 V617F )有密切联系。 JAK2 介导的信号转导,在促进或调节细胞的增殖中起重要作用。研究证实大部分PV及部分ET、PMF患者存在 JAK2 基因第14个外显子点突变,即 JAK2 基因编码序列第617号位氨基酸的第一位碱基发生G-T突变( JAK2 V617F )。PV患者这一基因突变的检出率为65%~97%。 JAK2 V617F 突变还可能通过JAK-STAT信号传导通路使红系祖细胞生成蛋白bcl-xl过表达;bcl-2 或 bcl-xl 抗凋亡蛋白的上调是维持红系祖细胞生存的重要机制。 JAK2 基因12号外显子突变见于50%~80%的 JAK2 V617F 突变阴性的PV患者。 JAK2 基因12号外显子突变包括缺失、点突变等多种类型,以 K539L、N542-543del、E543-544del最为常见,主要为杂合突变。与 JAK2 V617F 阳性的PV患者相比,第12号外显子突变患者表现出更高的血红蛋白水平,血小板及白细胞计数较低,但两者在血栓事件发生情况、向白血病及继发性骨髓纤维化转化风险及死亡发生率等方面无显著差异。
虽然 JAK2 基因突变在PV发病机制中有重要作用,其他基因突变在PV发病中的作用也不容忽视。PV患者中NF-E2 及PTP-MEG2过表达与EPO非依赖性红系集落形成呈正相关。PV中30%~40%可见染色体异常,9p染色体杂合性缺失(LOH)大约占33%。这是至今为止发现的最常见的染色体异常之一。常见的异常染色体核型还包括del(20q)及8号和9号染色体异常,其他有del(13q)及dup(1q)、del(5q)等。染色体异常和基因突变导致酪氨酸磷酸酶活性改变是PV发病主要机制。
真性红细胞增多症的主要病理生理基础是红细胞过度增生引起全血容量增多和血黏滞度增高,导致全身血管扩张和血流缓慢,可引起血管栓塞,以静脉血栓较多见。出血系由于血管扩张充血、血管内皮损伤和血小板功能异常引起。
【临床表现】
起病隐匿,偶在血常规检查时发现,也可因血栓形成及出血症状而就诊。主要临床表现有以下几个方面:
(一)血管神经系统的表现
早期有头痛、头昏、头胀、耳鸣、眩晕、健忘、肢体麻木、出汗等。重者可出现盲点、复视和视力模糊等症状;也可有心绞痛、间歇性跛行。红斑性肢痛多发生在下肢。
(二)血栓形成和栓塞症状
可发生在脑动脉、冠状动脉和外周动脉,引起脑血栓、心肌梗死等严重后果。血栓性静脉炎主要发生肺部;肠系膜、肝、脾和门静脉也可发生,引起相应器官的症状,如巴德-吉(基)亚利综合征(Budd-Chiari syndromes)。
(三)出血症状
常见鼻出血、牙龈出血和皮肤黏膜瘀点、瘀斑等。
(四)高代谢和组胺增高表现
易发高尿酸血症、痛风。消化性溃疡发生率较正常人高4~5倍,可引起消化道出血。皮肤 瘙痒也常见,10%可伴荨麻疹。
常见特征是面部、鼻、耳、唇、手掌和结膜充血,呈暗红色,如醉酒样。球结膜和口腔充血。3/4的患者有脾大,2/3有肝大,1/3有高血压,以收缩压升高明显。
【实验室检查】
(一)血常规
红细胞数大多在6×10 12 /L~10×10 12 /L,血红蛋白多在170~240g/L,血细胞比容为55%~80%,红细胞形态多数正常或轻度大小不一,偶见幼红细胞。有明显出血或多次放血者,红细胞可为低色素、小细胞性。白细胞数可轻度升高,半数患者血小板增多达450×10 12 /L~1000×10 9 /L,个别更高,可见巨型血小板,偶见巨核细胞碎片。
(二)骨髓象
增生活跃或明显活跃,粒、红、巨核细胞显著增生,尤其以幼红细胞为甚。粒系中以中性晚幼粒及杆状核细胞多见。巨核细胞增多,形态较大。骨髓细胞外铁和铁粒幼细胞减少或消失。骨髓切片显示粒、红、巨核三系细胞增生,脂肪细胞被造血细胞代替。合并骨髓纤维化时网状纤维增加。
(三)血容量及理化特性
用核素标记法测定红细胞容量增多(男性>36ml/kg,女性>32ml/kg),为重要的实验诊断依据。全血容量增加,血浆容量正常。血液比重高达1.070~1.080。血黏滞度为正常值的5~8倍。
(四)动脉血氧饱和度及促红细胞生成素
结果均在正常范围。由于粒细胞和血小板计数均增高,动脉血氧应及时测定,否则造成低氧血症的假阳性。
(五)血液生化
血及尿中促红细胞生成素(EPO)水平往往降低,但少数患者也可以正常。多数患者尿酸增加,血清维生素B 12 及维生素B 12 结合力增加。2/3患者高组胺血和尿血清。γ球蛋白可增多,α 2 球蛋白降低。
(六)染色体及基因
染色体异常发生率30%~40%,9p LOH,del(20q)及8号和9号染色体三体,del(13q)及dup(1q)、del(5q)等。 JAK2 V617F 点突变对PV有极高的诊断价值。90%~95%的患者可以检测到14号外显子上的 JAK2 V617F 基因突变,另有部分患者可以检测到 JAK2 基因第12号外显子突变。
(七)其他
红系祖细胞培养不加EPO可有红系爆式集落形成单位(BFU-E)、红系集落形成单位(CFU-E)。促红细胞生成素受体对促红细胞生成素表现为低亲和性,无促红细胞生成素基因突变。嗜中性粒细胞碱性磷酸酶活性增高。少数病例有血小板聚集、黏附功能不佳,血小板第3因子活力降低。
【诊断和鉴别诊断】
(一)PV的诊断标准
根据WHO2016年髓系肿瘤标准,PV的诊断标准有所变更,具体如下:
1.主要标准
(1)Hb>185g/L(男)或>160g/L(女);或红细胞压积:男性>49%,女性>48%;或红细胞量增加。
(2)骨髓活检示年龄校正三系血细胞增生(全髓增生),即显著红系、粒系和巨核系增殖。
(3) JAK2 V617F (+);或 JAK2 外显子12突变阳性。
2.次要标准
血清促红细胞生成素低于正常参考范围
(二)真性红细胞增多后骨髓纤维化(post-PV MF)诊断标准
1.主要标准
以下2条均需满足:①此前按WHO诊断标准诊断为PV;②骨髓活检显示纤维组织分级为2/3级(按0~3级标准)或3/4级(按1~4级标准)。
2.次要标准
至少符合其中2条:①贫血或不需要持续静脉放血(在未进行降细胞治疗情况下)或降细胞治疗来控制红细胞增多;②外周血出现幼稚粒细胞、幼稚红细胞:③进行性脾大(此前有脾大这超过左肋缘下5cm或新出现可触及的脾大);④以下3项体质性症状中至少出现1项:过去6个月内体重下降>10%,盗汗,不能解释的发热(>37.5℃)。
符合3项主要标准,2项主要标准加1项次要标准,即可诊断真性红细胞增多后骨髓纤维化。
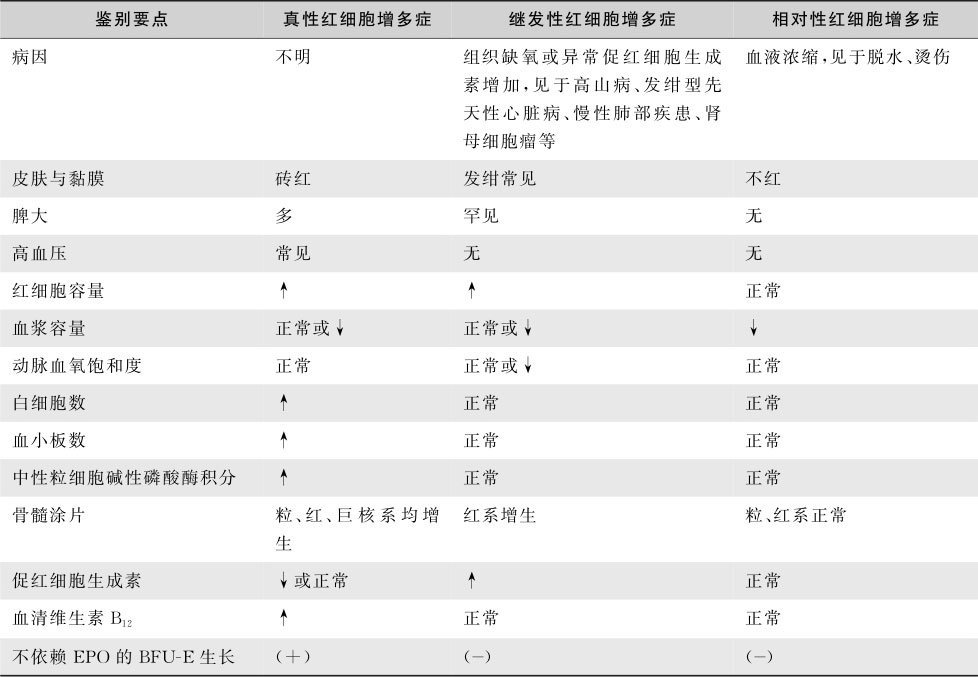
红细胞增多症尚可继发于下列情况:①组织缺氧引起促红细胞生成素增加,如有右至左分流的先天心脏病、慢性肺部疾患、高铁血红蛋白血症等;②促红细胞生成素或促红细胞生成素样物质异常增多引起红细胞增多症,如肾母细胞瘤、肝癌等。真性红细胞增多症与继发性及相对红细胞增多相鉴别,参见表16-4-1。此外,中年患者体型肥胖、神经质、轻度高血压,发生一过性红细胞增多者称为应激性红细胞增多症。
表16-4-1 各种红细胞增多症的鉴别要点
【治疗】
(一)治疗目标
PV的治疗目标是避免血栓形成,控制疾病相关症状,预防post-PV MF和(或)急性白血病转化,多血症期治疗目标是控制HCT<45%。
(二)一线治疗
1.预防血栓
由于栓塞是PV患者的主要死亡原因,因此,确诊患者均应进行血栓预防。首选口服低剂量阿司匹林(100mg/d),不能耐受者可选用双嘧达莫。
2.静脉放血
疗效迅速,简单安全。定期的放血治疗保持红细胞数量在正常范围,保持男性血红蛋白≤140g/L,女性血红蛋白≤120g/L,可以避免血栓形成的并发症;每周静脉放血2~3次,每次300~500ml,老年及有心血管疾病放血应慎重,每次不超过200~300ml。一旦形成缺铁状态,放血治疗可间隔3个月1次。有条件使用血细胞分离机,可单采红细胞,但应补充和单采红细胞等容积的代血浆或同型血浆。放血仅减少红细胞,不能抑制骨髓增生;单纯的放血有较高的出血和血栓形成的危险,故放血治疗同时采用骨髓抑制药物更为合适。
3.降细胞治疗
有效率为80%~85%,适用于血细胞显著增多,反复放血无效者。羟基脲对真性红细胞增多症骨髓抑制较好,且一般无致白血病作用。常用剂量为15~20mg/kg,维持白细胞在3.5~5.0×10 9 /L时须间歇服用。羟基脲无效者可选用白消安,2~6mg/d;苯丁酸氮芥(CB1348)4~10mg/d;环磷酰胺100~150mg/d,左旋苯丙酸氮介(马法仑)4~6mg/d;烷化剂有致突变作用。哌酰溴烷(Pipobroman)开始剂量每日75mg,当血细胞比容下降改为每日50mg,维持量25mg,每日1次。本药属烷化剂类,毒性较其他烷化剂低。三尖杉碱对红系DNA合成有抑制作用,国内报告疗效满意,疗效持续时间也较长。
重组α干扰素作用机理是抑制造血细胞的增殖作用,同时可以抑制血小板衍生生长因子(PDGF)以减少骨髓纤维组织增生。剂量每次300万U~500万U,皮下注射,每周3次。用药6~12个月后70%的患者TCT获得控制。20%的患者部分缓解,10%无效。此外,干扰素治疗还可以使血小板计数升高、皮肤瘙痒和脾大得到改善。
(三)二线治疗选择
1.放射核素 32 P
32 P通过释放β射线,直接阻止骨髓造血细胞核分裂,抑制造血。静脉注射首剂量3~5mCi,2~3个月后,血常规可恢复正常,肝脾缩小,12~16周后,需第二次静脉给药2~3mCi。口服剂量4.5~8mCi,分2次口服,隔1周1次。给药前后给低磷饮食2~4周。本法应用方便,疗效高,缓解率达75%~85%,缓解期可持续半年到数年。因可造成骨髓造血抑制,急性白血病和非造血系统肿瘤发生率提高;因此仅用于需经常放血,长期应用骨髓抑制药不见效,以及肝、肾功能尚好的老年患者。
2.白消安及其他药物
羟基脲无效者可选用白消安,2~6mg/d;苯丁酸氮芥(CB1348)4~10mg/d;环磷酰胺100~150mg/d,左旋苯丙酸氮介(马法仑)4~6mg/d;烷化剂有致突变作用。哌酰溴烷(pipobroman)开始剂量每日75mg,当血细胞比容下降改为每日50mg,维持量25mg,每日1次。本药属烷化剂类,毒性较其他烷化剂低。三尖杉碱对红系DNA合成有抑制作用,国内报告疗效满意,疗效持续时间也较长。
3.阿那格雷(Anagrelide)
用于真性红细胞增多症伴血小板增多而用羟基脲不能控制的患者。用法见本章第三节“原发性血小板增多症”。
4.芦可替尼
2014年12月芦可替尼被FDA批准用于羟基脲疗效不佳或不能耐受的PV患者。推荐其实剂量为20mg/d,在开始治疗的前4周不进行剂量调整,每次剂量调整间隔不应少于2周,最大剂量不超过50mg/d。芦可替尼最常见的血液学不良反应为3/4级的贫血、血小板减少及中性粒细胞减少,但极少因此导致中断治疗。
(四)其他
有高尿酸血症应口服别嘌醇及碱性药物。瘙痒者用组胺H 2 受体拮抗剂如西米替丁,赛庚定。红斑性肢痛可用小剂量阿司匹林,无效也可用阿那格雷。PV后骨髓纤维化期伴巨脾者,各种治疗无效,亦可考虑切脾,但务必慎重。有报道用格列卫(STI571)400mg/d,口服,可以减少放血治疗次数。
【预后】
本病进展缓慢,如无严重并发症可生存10~15年以上。不治疗者平均生存仅18个月。死亡原因主要为血栓、栓塞及出血,部分患者晚期可转变为白血病或发生骨髓纤维化、骨髓衰竭。
主要参考文献
1.徐俊卿,徐哲锋,王静雅,等.615例Ph染色体BCR-ABL融合基因阴性骨髓增殖性肿瘤患者的症状负荷评估.中华血液学杂志,2016,37(1):26-29.
2.肖志坚.骨髓增殖性肿瘤和骨髓增生异常综合征/骨髓增殖性肿瘤:开启分子诊断新时代.中华血液学杂志,2014,35(5):385-386.
3.Bench AL,White HE,Foroni L,et al.Molecular diagnosis of the myeloproliferative neoplasms:UK gidelines for the detection of JAK2 V617F and other relevant mutations.Br J Hematol,2013,160(1):25-34.
4.Barhui T,Thiele J,Carobbio A,et al.Masked polycythemia vera diagnosed according to WHO and BCSH classification.Am J Hematol,2014,89(2):199-202.
5.Barhui T,Thiele J,Vannucchi AM,et al.Rethinking the diagnostic criteria of polycythemia vera.Leukemia,2014,28.(6):1191-1195.
第三节
原发性血小板增多症
原发性血小板增多症(Essential thrombocythemia,ET)系主要累及巨核细胞系的MPN。其特征为外周血中血小板持续增多,且伴功能异常,骨髓中巨核细胞过度增殖,临床有自发出血倾向及或有血栓形成,约半数患者有脾大。
【病因及发病机制】
本病是起源于多能干细胞的克隆性疾病,巨核细胞-血小板系列占优势增殖。可能是因为异常克隆对调节因子的优先反应使得其能够分化为成巨核细胞-血小板系列。这些改变还伴随着异常CFU-MEG克隆细胞核的核内复制。其他的细胞因子(例如IL-3,IL-6,IL-11)也在不同的阶段起作用,并与促血小板生成素起协同作用。近来研究发现,原发性血小板增多症中存在 JAK2 基因点突变即 JAK2 V617F突变,该突变发生率为23%~57%。 JAK2 基因突变可引起JAK-STAT等信号系统激活外,还可引起血小板生成素受体(TPOR)、粒细胞集落因子受体(G-CSFR)等异常活化及信号传导,造成巨核细胞系及粒系祖细胞异常增生,可能是ET的发病原因。
最近,应用全基因组测序技术在67%~88%的 JAK2 、 MPL 突变阴性的ET或PMF患者可检出钙网蛋白(calreticulin,CALR)基因9号外显子的缺失突变,且与 JAK2 、 MPL 突变互排,提示CALR突变是MPN重要的驱动突变。钙网蛋白在人类肿瘤肿瘤的免疫逃逸,未折叠蛋白反应(unfolded protein response,UPR)和Ca 2+ 信号传递中发挥着重要的作用。目前已有超过50种CALR突变类型被报道,最常见的突变类型为9号外显子上52对碱基的缺失(p.L367fs*46,1型)和5对碱基TTGTC的插入(p.K385fs*47,2型),大约占CALR总突变率的90%。
【临床表现】
原发性血小板增多症,每年发病率为0.1/10万人。中位发病年龄60岁(范围2~90岁),好发于50~70岁。女:男=1.3∶1。起病缓慢。约有20%的患者,尤其年轻患者,发病时无症状,偶尔因血小板增多及脾大进一步检查而确诊。1/3患者就诊时表现功能性或血管舒缩性症状包括血管性头痛、头昏、视觉模糊、手掌及足底灼痛感,肢体末梢麻木。80%患者因原因不明的出血及血栓形成而就诊。出血为自发性,可反复发作,以胃肠道出血常见,也可有鼻及齿龈出血、血尿、呼吸道出血、皮肤及黏膜淤斑,但紫癜少见。有时可因手术后出血不止而被发现。偶有脑出血,引起死亡。血栓发生率较出血少。国外报告血栓形成较国内多见。国内统计30%有动脉或静脉血栓形成。静脉以脾、肠系膜及下肢静脉为血栓好发部位。下肢血管栓塞后,可表现肢体麻木感、疼痛、甚至坏疽。也有表现为红斑性肢痛,间歇性跛行。肠系膜血管血栓形成可致呕吐、腹痛。肺、肾、肾上腺或脑内发生栓塞可引起相应临床症状,可成为致死的原因。脾大见于80%以上的病例,一般为轻到中度肿大,少数患者有肝大。
【实验室检查】
(一)血常规
血小板计数多在1000×10 9 /L~3000×10 9 /L之间,最高可达20 000×10 9 /L。血小板形态一般正常,但有巨大型、小型及畸形,常聚集成堆,偶尔见到巨核细胞碎片及裸核。白细胞计数可正常或增高,多在(10~30)×10 9 /L,偶尔可达到(40~50)×10 9 /L,一般不超过50×10 9 /L,分类以中性分叶核粒细胞增多为主。因失血少数患者可致低色素性贫血,红细胞大小不均、中心淡染、多染性、可见嗜碱性点彩及豪-胶小体。
(二)骨髓象
有核细胞增生活跃或明显活跃,巨核细胞增生尤为显著,原始及幼稚巨核增多,有大量血小板聚集成堆。
(三)出、凝血试验
出血时间延长,凝血酶原消耗时间缩短,血块退缩不良。血小板黏附功能及肾上腺素和ADP诱导的聚集功能均降低,但对胶原聚集反应一般正常。凝血酶原时间正常或延长,白陶土部分凝血活酶时间延长。
(四)生化
血尿酸、乳酸脱氢酶、血清酸性磷酸酶均增高,中性粒细胞碱性磷酸酶活性也增高。部分患者因血小板破坏,大量钾离子释放到血中,引起假性高钾血症。
(五)其他
染色体检查部分患者有21号染色体长臂缺失(21q - ),也有报告21号染色体长臂大小不一的变异。骨髓祖细胞培养有自发的巨核细胞或红细胞克隆形成。23%~57%患者有 JAK2 基因突变。
【诊断和鉴别诊断】
(一)2016年WHO提出的诊断标准
1.主要标准
(1)血小板持续≥450×10 9 /L。
(2)骨髓活检示主要为巨核系增生,多为体积大、成熟巨核细胞,无明显粒系或红系增生和左移。
(3)不符合 WHO关于 BCR-ABL1 阳性CML,PV、PMF、骨髓增生异常综合征或其他髓系肿瘤的诊断标准。
(4)有 JAK2 、 CALR 或 MPL 突变阳性。
2.次要标准
克隆性标志物阳性或反应性血小板增多阴性(应排除反应性血小板增多症如:缺铁、切脾后、感染、炎症、结缔组织病、肿瘤转移、淋巴增殖性疾病、手术后)。
诊断时需要满足全部4项主要标准或前3项主要标准加1项次要标准。
(二)鉴别诊断
1.其他骨髓增殖性肿瘤
真性红细胞增多症、慢性粒细胞白血病及骨髓纤维化,皆可伴有血小板增多。但真性红细胞增多症以红细胞增多为突出表现。 JAK2V617F 阳性的ET更类似于PV,其血液红细胞和白细胞水平较高,骨髓增生更明显,静脉血栓形成及转化为PV的可能更大,故有人提出 JAK2 V617F阳性的ET和PV甚难区别。 JAK2 V617F阴性的ET常有脾脏肿大,骨髓巨核细胞发育不良,容易转化为白血病。慢性粒细胞白血病以粒细胞系列增生为主,血中白细胞显著增多,出现幼稚粒细胞,中性粒细胞碱性磷酸酶积分明显降低,染色体检查可见到Ph染色体。骨髓纤维化的患者外周血中有幼红、幼粒细胞,红细胞大小不等及见到泪滴样红细胞增多,骨髓大多干抽,骨髓活检有纤维化的表现。原发性铁粒幼细胞贫血及骨髓增生异常综合征(5q-综合征)也可有血小板增多,但有相应的细胞学特点。
2.继发性血小板增多症
见于脾切除后、脾萎缩、急性或慢性失血、溶血、外伤及手术后,慢性感染、风湿性疾病、坏死性肉芽肿、炎症性肠病、恶性肿瘤、分娩、应用肾上腺类等药物,戒酒后、维生素B 12 和叶酸缺乏纠正后也可引起血小板增多。骨髓细胞培养,原发性血小板增多症有自发性巨核细胞集落形成,可与继发性区别。
【治疗】
ET治疗的目的主要是减少血小板数量,预防血栓和出血的发生。治疗方案根据ET患者发生血栓并发症的危险分度分级而制定。根据年龄、有无心血管疾病危险因素、血栓史及血小板计数分为低、中、高三级:
低危:<40岁,无心血管疾病危险因素,如果血小板<1500×10 9 /L可以观察而不治疗或给予小剂量阿司匹林;
中危:40~60岁,无心血管危险因素,小剂量阿司匹林治疗;
高危:年龄>60岁,有血栓症既往史,或血小板>1500×10 9 /L和(或)有心血管危险因素,抑制细胞治疗和小剂量阿司匹林。但必须强调的是这些低危的患者中也有较少比例的栓塞发生率。
(一)抗血小板治疗
小剂量阿司匹林(100mg/d)若患者不能耐受或阿司匹林使用禁忌证,可使用氯吡格雷抗血小板治疗。其次双嘧达莫、吲哚美辛有防止血小板聚集作用,在阿司匹林不能耐受时可以选用。有血栓形成者用肝素或双香豆素类抗凝。
(二)骨髓抑制性药物
1.羟基脲
对于高危ET常常选用,剂量1~2g/d,分2~3次口服。白消安也可选用,开始4~6mg/d,分次或一次口服,待血小板减少到一半时,剂量也相应减少一半。血小板减少至正常时停药或改为维持量。其他如瘤可宁和环磷酰胺等均可按病情或个体敏感性分别选用。
2.阿那格雷(anagrelide)
为环磷腺苷二酯酶抑制剂。可抑制巨核细胞成熟,使血小板产生减少。有效率90%,目前是可供选择的一线药物。推荐起始剂量每次0.5mg,每天2次,至少1周后开始调整剂量,维持血小板<600×10 9 /L。剂量增加每周不超过0.5mg/d,最大单次剂量为2.5mg,每日最大剂量为10mg,维持剂量2.0~2.5mg/d。不良反应有头痛、液体潴留、直立性低血压、心悸、心动过速、心力衰竭等。
3.干扰素-α
干扰素-α可抑制巨核细胞生成血小板及使血小板生存时间缩短,剂量每天1次,每次300万U,根据耐受性和治疗反应调整剂量,每周3次的维持量可抑制血小板生成达数年之久。停药后血小板回升。但因长期应用的相关副作用,不推荐作为一线用药。
(三)血小板分离术
可以迅速减少血小板数量,改善症状。常用于胃肠道出血,妊娠及分娩、选择性手术前。
(四)放射性核素磷( 32 P)
可口服或静脉注射,首次剂量3~4mCi,如有必要3个月后再给药一次。现一般不主张用,因有诱发白血病的可能。
(五)其他
切脾是禁忌的,因术后可致血小板明显增多,血栓形成。
【病程预后】
根据血小板增多的程度,病程不一。大多数病例进展缓慢,中位生存期10~15年。约25%患者可转为骨髓纤维化,部分病例可转化为真性红细胞增多症,<5%病例可转化为MDS或AML。重要脏器有血栓形成及出血,常为本症致死的主要原因。
主要参考文献
1.Gugfiemelli P,Nangalia J,Green AR,et al.CALR mutations in myelopro-liferative neoplasms:hidden behind the retieulum.Am J Hematol,2014,89(5):453-456.
2.Passamonti F,Caramazza D,Maffioli M.JAK inhibitor in CALR-mutant Myelofibrosis.N Engl J Med,2014,370(12):1168-1169.
3.Rumi E,Pietra D,Ferretti V,et a1.JAK2 or CALR mutation status deftnes subtypes of essential thrombocythemiaith substantially difierent clinical course and outcomes.Blood,2014,123(10):1544-1551.
4.Mesa R,Miller CB,Thyne M,et al.Myeloproliferative neoplasms(MPNs)have a significant impact on patients'overall health and productivity:the MPN Landmark survey.BMC Cancer,2016,16:167 DOI 10.1186.
5.Tefferi A,Lasho TL,Finke CM,et al.CALR vs JAK2 vs MPL-mutated or triple-negative myelofibrosis clinical cytogenetic and molecular comparisons.Leukemia,2014,28(7):1472-1477.
6.Rumi E,Pietra D,Ferretti V,et al.JAK2 or CALR mutation status defines of essential thromboeythemia with substantially Different clinical course and outcomes.Blood.2014,123(10):1544-1551.
第四节
原发性骨髓纤维化
原发性骨髓纤维化(primary myelofibrosis,PMF)简称“骨纤”,是MPN的一种,主要表现为骨髓中巨核细胞和粒细胞显著增生伴反应性纤维结缔组织沉积,伴髓外造血。临床特点起病缓慢,脾脏常明显肿大,外周血中出现幼红和幼粒细胞,骨髓穿刺常干抽和骨髓增生低下。男和女发病率相等,白种人较其他种族多见。好发于中老年,但儿童甚至婴儿亦可见到。
【发病机制】
本病系多能干细胞恶性克隆性疾病。偶尔有非随机的染色体异常例如20q-,13q-,和1q三体,骨髓纤维化的程度和髓外造血的范围并不相关。近年发现一组与结缔组织增生有关的生长因子如血小板衍生生长因子(PDGF),巨核细胞衍生生长因子(MKDGF)、上皮生长因子(EGF)及β转化生长因子(β-TGF)在巨核细胞中合成,储存在血小板的α颗粒中。PMF有无效性巨核细胞生成,破坏的巨核细胞释放出大量PDGF、EGF及β-TGF等协同刺激纤维细胞的增生,分泌胶原,同时释放出血小板因子。后者抑制胶原酶的活性,使胶原降解减少,导致骨纤的形成。但在急性白血病和播散性恶性肿瘤的骨髓中发生纤维化,巨核细胞并不增多,可能系恶性肿瘤释放出类似的生长因子而引起纤维组织增生。临床和实验室研究证实维生素D及甲状旁腺素的代谢产物能调节骨髓内胶原的沉积。因而当甲状旁腺功能或维生素D代谢紊乱时,也可导致骨髓纤维化。研究表明原发性骨髓纤维化与 JAK2 ( JAK2 V617F)突变有密切联系。此外约10% JAK2 V617F阴性的PMF患者中有 TPO 基因激活突变及慢性骨髓增生性白血病病毒癌基因( MPL )突变( MPLW515L / K )与PMF发生也可能有关。最近全基因测序技术发现,在 JAK2 突变阴性的PMF中有67%~88%的可检出 CALR 基因9号外显子的缺失突变,且与 JAK2 、 MPL 突变互排,提示 CALR 突变是MPN重要的驱动突变。
至于髓外造血也是同一异常刺激引起的增生反应;也可能由于骨髓纤维化过度增生破坏正常的骨髓超微结构,因而造血前体细胞从骨髓中释放进入周围血,并在肝、脾等髓外器官增殖,而不是代偿作用。
【临床表现】
大多在50~70岁间发病。起病缓慢,约30%的患者诊断时无自觉症状或仅表现有乏力、多汗、消瘦、体重减轻及脾大引起上腹闷胀感等。严重的患者可有骨痛、发热、贫血、出血,因高尿酸血症有4%的患者可引起肾结石及40%引起痛风性关节炎。个别患者因耳骨硬化可致听力减退。发热多数可由感染引起,可有原因不明腹泻。由于髓外造血可引起相应器官的症状,几乎所有患者均有脾大,有的报道脾大速度每年1cm,约50%患者就诊时脾大已达盆腔,质地中等硬。脾不大者罕见。脾大与脾血流量增加,肝内血流阻力增高及脾髓外造血有关。50%~70%的患者有肝大,多为轻到中度肿大,个别的可达脐下,质坚硬而不痛,表面光滑。约有10%~20%的病例合并肝硬化,由于肝血窦周围血管阻塞及肝窦髓外造血引起门静脉血流量增加所致。因肝静脉或门静脉内血栓形成可导致门静脉高压或Budd-Chiari综合征。胸骨压痛少见。面色苍白和贫血程度有关。少数患者由于无效红细胞生成可有黄疸。因为淋巴结极少有髓外造血灶,故肿大不明显。
【实验室检查】
(一)血常规
大多数患者就诊时均有轻重不等的贫血,晚期可有严重的贫血,通常属于正细胞、正色素型。成熟红细胞有显著泪滴样改变及异形。贫血的原因可由于脾大、脾功能亢进,红细胞破坏增加部分患者长期大量红细胞增生,可继发叶酸缺乏,此外血容量相对增多,以及红细胞无效生成。网织红细胞轻度增多,在2%~5%。约70%患者外周血中出现幼粒、幼红细胞也是本病的特征之一。
白细胞计数多增加,一般在(10~30)×10 9 /L,很少超过50×10 9 /L,少数患者白细胞可减少到(2~4)×10 9 /L。分类中以成熟中性粒细胞为主,也可见到中幼粒、晚幼粒细胞,甚至原粒细胞和早幼粒细胞。嗜酸性粒细胞和嗜硷性粒细胞轻度增加。
血小板计数高低不一,约1/3病例血小板增加,个别可达1000×10 9 /L。外周血中可见到大而畸形的血小板,偶见巨核细胞碎片或巨核细胞。血小板功能有缺陷。
(二)骨髓穿刺涂片及活检
骨髓穿刺约有1/3的病例“干抽”现象。骨髓涂片有核细胞常增生低下,也可为增生象。骨髓活检见到大量网状纤维组织为诊断本病的依据。根据骨髓中保留的造血组织和纤维组织增生的程度不同,骨髓病理改变可分为三期:①早期全血细胞增生期伴有纤维组织增生;②中期骨髓萎缩与纤维化期;③晚期骨髓纤维化和骨质硬化期。
(三)脾穿刺液涂片
脾穿刺涂片显示淋巴细胞和粒、红、巨核三系细胞均增生。脾穿刺涂片诊断价值较大,但有出血的危险,必须慎重考虑。
(四)肝穿刺与活检
如同脾脏一样有髓外造血。在肝窦中见到幼稚红细胞及巨核细胞;幼稚粒细胞在门脉区多见。
(五)X线检查
约50%的病例X线检查有骨质硬化征象,骨质密度不均匀性增加,伴有斑点状透亮区,形成所谓“毛玻璃”样改变;也可见到骨质疏松,新骨形成及骨膜花边样增厚。骨质变化好发于长骨的干骺端、脊椎、骨盆、下肢的长骨、肱骨、肋骨等尤为明显,部分病例也有颅骨变化。
(六)放射性核素骨髓扫描
放射性胶体( 99 锝、 52 铁、 111 铟等)为骨内红髓、脾、肝等扫描摄取而出现放射性浓集区。骨髓纤维化的患者肝、脾髓外造血区积累了大量放射核素,长骨近端等有纤维组织增生改变的红髓则不能显示放射浓集区。
(七)染色体检测及基因检测
约半数染色体不正常,常见的为C组染色体呈三体,也可有del(13q),del(20q)。未见到Ph染色体。50%~60%的PMF患者可检出 JAK2 基因14号外显子突变, MPL 基因突变可见于8%~10%的PMF患者,67%~88%的 JAK2 突变阴性PMF患者可检出 CALR 基因9号外显子的缺失突变,且与 JAK2 、 MPL 突变互排。
(八)其他
血清碱性磷酸酶、尿酸、乳酸脱氢酶、维生素B 12 及组织胺均见增高。2/3的慢性病例血清碱性磷酸酶因骨病改变增加。但随着病程进展逐渐降低。
【诊断及危险度分级】
(一)诊断标准
2016年WHO髓系肿瘤分类标准,把原发性骨髓纤维化分为纤维前/早期原发性骨髓纤维化(prePMF)和显性原发性骨髓纤维化(PMF)两期。
1.prePMF诊断标准
诊断时需满足3项主要标准和至少1项次要标准。
(1)主要标准:
1)巨核细胞增生和不典型性,网状纤维≤1度,伴骨髓细胞增殖,常见红细胞生成功能障碍。
2)不符合 WHO关于 BCR-ABL1 阳性CML、PV、PMF,骨髓增生异常综合征或其他髓系肿瘤的诊断标准。
3) JAK2 , CALR 或 MPL 基因突变阳性,或基因突变阴性,而其他克隆性标志物阳性,或轻度反应性骨髓网状纤维化阴性。
(2)次要标准:
表现为下列至少1项,且经2次连续检查证实。
1)非共患疾病所引起的贫血。
2)白血病增多(≥11×10 9 /L)。
3)脾明显肿大。
4)乳酸脱氢酶(LDH)高于正常水平上限。
2.显性PMF诊断标准
诊断时需要满足全部3项主要标准和至少1项次要标准。
(1)主要标准:
1)巨核细胞增生和不典型性,伴网状和(或)2度或3度胶原纤维增生。
2)不符合 WHO关于 BCR-ABL1 (+)CML、PV、骨髓增生异常综合征或其他髓系肿瘤。
3)有 JAK2 、 CALR 或 MPL 基因突变阳性,或基因突变阴性,而其他克隆标志物阳性,或轻度反应性骨髓网状纤维化阴性(无感染、自身免疫性疾病或其他慢性炎症性疾病、毛细胞白血病或其他淋巴系肿瘤、转移性肿瘤或慢性中毒性骨髓病)。
(2)次要标准:
表现为下列至少1项,且经2次连续检查证实。
1)非共患病所引起的贫血。
2)白细胞增多(≥11×10 9 /L)。
3)脾明显肿大。
4)乳酸脱氢酶(LDH)高于正常水平上限。
5)骨髓病性贫血。
(二)危险度分级
PMF患者根据国际预后评分系统(IPSS)进行危险度分层。该系统包括5个危险因素:年龄>65岁,全身症状(诊断前1年体重下降>10%、不明原因的发热、严重盗汗超过1个月)、Hb<100g/L,WBC>25×10 9 /L、外周血原始细胞≥1%。每一项积1分。低危:0分,中位生成期11.3年,中危-1:积分2分,中位生存时间7.9年;中危-2:2分,中位生存时间4年;高危:≥3分,中位生存时间2~3年。
【鉴别诊断】
PMF应和下列疾病鉴别:①慢性粒细胞白血病:两者均可巨脾,白血病计数增加,周围血出现中幼粒、晚幼粒细胞,但慢粒发病年龄轻,白血病计数常超过100×10 9 /L~300×10 9 /L,血片中较少有幼红细胞,红细胞畸形也不似PMF典型。粒细胞碱性磷酸酶活性降低或消失以及有Ph染色体均可与PMF区别。②本病尚须与低增生性白血病以及引起幼粒-幼红细胞贫血的其他疾病相区别。 JAK2 、 CALR 及 MPL 基因突变阳性可与继发性骨髓纤维化鉴别,后者可从临床表现或特殊检查中获得帮助。有时多部位、多次骨髓穿刺及活检,才能除外继发性骨纤。
【治疗】
目前对骨髓纤维化的治疗缺少特效的措施。治疗应根
(一)纠正贫血
雄激素及蛋白合成剂有改善骨髓造血功能的作用。约有50%的患者对雄激素有较好的疗效,部分的患者还可使白细胞和血小板增多;肝病者慎用。丙酸睾酮的剂量为100mg,隔日肌内注射,也可用长效的庚酸睾酮100~400mg,每周1次,肌内注射,或口服司坦唑醇、羟基雄酮、羟甲雄烯异
 唑等。用法参阅“第二章第二节“再生障碍性贫血和其他骨髓衰竭综合征”。上述药物有时需用3~4个月以上才能见效。合并有溶血或血清中找到免疫复合物或自身抗体者,可给泼尼松治疗,剂量为20~30mg/d。
唑等。用法参阅“第二章第二节“再生障碍性贫血和其他骨髓衰竭综合征”。上述药物有时需用3~4个月以上才能见效。合并有溶血或血清中找到免疫复合物或自身抗体者,可给泼尼松治疗,剂量为20~30mg/d。
(二)细胞毒药物治疗
可以抑制骨髓造血组织的异常增殖,同时可以抑制免疫发病机制;从而防止骨髓纤维组织的进一步发展。一般用于脾大,骨髓处于增生阶段,周围血细胞稍多的病例。常用的有①苯丁酸氮芥联合泼尼松:苯丁酸氮芥每日15mg,泼尼松每日30mg。3~4周1个疗程,可以保持血红蛋白较好的水平和减轻脾大。②羟基脲:开始剂量是500mg,每日1次,渐加量至每日用量1000~2000mg。密切观察脾大和白细胞、血小板数。少数患者用药1年内可以改善症状。③马利兰或6-巯基嘌呤(6-TG):马利兰剂量每日2~4mg,6-TG每日20~40mg,6~9周后肝、脾可缩小,血红蛋白增加。
2011年11月FDA批准第一个JAK抑制剂芦可替尼(ruxolitinib)用于IPSS中危-2和高危组PMF患者的治疗。英国骨髓纤维化研究和诊治指南(2014)推荐骨髓纤维化(包括PMF、post-PV MF和post-ET MF)患者在以下情况下首选芦可替尼:①症状性脾大;②影响生活质量的骨髓纤维化相关症状;③骨髓纤维化导致的肝大和门脉高压。推荐的起始剂量依患者血小板计数:>200×10 9 /L,给予每次20mg,每天2次,(100~200)×10 9 /L为每次15mg,每天2次;(50~100)×10 9 /L。治疗过程中如血小板计数<50×10 9 /L,中性粒细胞绝对值<0.5×10 9 /L时应停药。停药应在7~10天内逐渐减停。停药过程中推荐加用泼尼松(20~30mg)。
(三)脾切除术
脾脏是本病主要髓外造血器官,约有10%~25%患者脾切除后可引起肝脏迅速肿大,血小板显著增高及感染的危险。因此,脾切除术一般仅限于:①巨脾有明显的压迫症状或出现脾梗死引起的持续性疼痛;②由于脾功能亢进引起顽固性溶血或血小板减少,经药物治疗无效且需长期反复输血但造血功能尚未完全丧失者;③伴有门静脉高压并发食管静脉曲张破裂出血者。对血小板数偏高者,术后容易发生静脉内血栓,一般视为手术禁忌证。晚期骨髓纤维化合并活动性肝病者,因手术后死亡率高达7.5%~25.7%,亦不应考虑脾切除术。
(四)脾区照射
对明显脾大者,照射后可使症状减据骨髓纤维组织增生的程度及临床表现,给予相应的措施。治疗的目的主要为改善骨髓的造血功能,纠正贫血、出血,缓解脾大所致的压迫症状。轻,脾脏缩小,但疗效短暂,4~6个月后又脾大,且有使周围血象进一步降低的副作用。
(五)干扰素-α
有抑制正常粒系祖细胞和巨核细胞增殖作用,常用α-2b干扰素治疗,但仅少数病例临床症状及体征取得一定程度的缓解。剂量为干扰素-α(300~500)万U皮下注射,每周3次。宜长期应用。
(六)1,25二羟维生素D 3
体外1,25二羟维生素D 3 可以抑制巨核细胞的增殖并诱导髓细胞向单核细胞及巨噬细胞转化,从而促使胶原纤维形成减少及裂解增加,剂量0.25~1.00μg/d。个别病例服药后血红蛋白及血小板数可有所增高,其确切疗效有待积累更多的临床资料。本药可引起血钙增高等不良反应,服药期间应定期随访测定血钙、血磷。
(七)造血干细胞移植
近来认为造血干细胞移植是有希望的治疗。同基因或异基因移植均可以改变骨髓纤维化的进程,而且存在移植物抗纤维化的作用。个别病例移植成功后骨髓纤维组织消失,且疗效不受纤维组织增生程度的影响。因移植相关的不良反应和患者的基础情况差及年龄较大等而不能广泛开展。非清髓性干细胞移植可以放宽移植的适应证。如果患者有不良的细胞遗传学(尤其是17号染色体)异常,或基本进展,或转为急性白血病,有条件者应尽快选择性异基因造血干细胞移植治疗。
(八)其他
有报告用大剂量甲泼尼龙治疗,可使贫血改善、脾缩小。开始剂量30mg/(kg·d),用4天,以后改为每日10、5、2、1mg/kg,各1周,继而口服泼尼松维持,渐减量停药。也有报道用免疫抑制剂:环孢素、硫唑嘌呤、大剂量免疫球蛋白静脉滴注可改善症状。另有报道沙利度安可改善血细胞减少状况,而TNF-α抑制剂(etanercept)可改善患者的一般情况。
【病程与预后】
病程长短不一,自1~20年不等,平均生存时间1~5年。国际预后评分系统(IPSS)低危:中位生存时间11.3年,中危-1:中位生存时间7.9年;中危-2:中位生存时间4年;高危:,中位生存时间2~3年。约有8%~20%的患者最后演变为急性白血病,死因多为严重感染、出血、心力衰竭。
主要参考文献
1.中华医学会血液分会白血病学组.原发性骨髓纤维化诊断与治疗中国专家共识(2015年版).中华血液学杂志,2015,36(9):721-725.
2.Verstovsek S,Mesa RA,Gotlib J,et a1.Efficacy,safety,and survival with ruxolitinib in patients with myelofibrosis:results of a median 3-year follow-up of COMFORT-I.Haematologica,2015,100(4):479-488.
3.Tefferi A.Guglielmerlli P,Lasho TL,et al.CALR and ASXL1 multations-based molecular prognositication in primary myelofibrosis:an international study of 570 patients.Leukemia,2014,28(7):1494-1500.
4.Rumi E Pietra D,ferretti V.et al.JAK2 or CALR mutation status defines of essential thromboeythemia with substantially Dif-ferent clinical course and outcomes Blood,2014,123(10):1544-1551.
5.欧阳苑,乔纯,王菊娟,等.BCR-ABL阴性 MPN患者CALR、JAK2及 MPL基因突变分析.中华医学杂志,2015,95(18):1369-1373.
第五节
嗜酸性粒细胞增多综合征
许小平
【概述】
外周血液中嗜酸性粒细胞占白细胞总数的1%~3%。嗜酸性粒细胞的绝对值上限为0.35×10 9 /L,高于此上限即可称为嗜酸性粒细胞增多(eosinophilia),并根据增多的程度将其分为:①轻度增多:嗜酸性粒细胞绝对值(0.351~1.500)×10 9 /L;②中度增多:>(1.5~5.0)×10 9 /L;③重度增多:>5.0×10 9 /L。临床上嗜酸性粒细胞增多可见于多种疾病,其中以寄生虫感染和变态反应性疾病为最常见。随病因不同临床表现多样。嗜酸性粒细胞起源于骨髓中的多向性髓系祖细胞,其发育、分化、成熟受IL-5、GM-CSF、IL-3等细胞生长因子的调控,一旦缺乏这些嗜酸性粒细胞生长因子,嗜酸性粒细胞将迅速凋亡。其中IL-5能特异性地促进其分化、发育、成熟和释放,在嗜酸性粒细胞生成增多中最为重要。正常情况下嗜酸性粒细胞主要驻留于组织中,特别是呼吸道、胃肠道和泌尿生殖道的上皮细胞和深层组织之间,在组织内其生存时间可达数周。嗜酸性粒细胞的形态学特征是核分两叶和胞质内有大量特殊的嗜酸性颗粒,决定了细胞的染色特性和功能特征。嗜酸性粒细胞的确切功能尚不明了,它参与免疫反应,能抵御大的不能被吞噬的病原体,如蠕虫类幼体。嗜酸性粒细胞具有晶体样核心,由多种蛋白组成,包括:①主要碱性蛋白(major basic protein,MBP),有MBP-1和 MBP-2,可破坏寄生虫和肿瘤细胞,对正常组织也有损害;②嗜酸性粒细胞阳离子蛋白(eosinophil cationic protein,ECP),可杀伤寄生虫,中和肝素,抑制淋巴细胞分裂;③嗜酸性粒细胞衍生神经毒素(eosinophil-derived neurotoxin,EDN),可损伤神经系统;④嗜酸性粒细胞过氧化物酶(eosinophil peroxidase,EPO),可对肿瘤细胞、寄生虫和细菌具有杀伤作用,对许多组织也有损伤作用。这些蛋白也可中和肝素的抗凝活性。嗜酸性粒细胞也能吞噬杀灭细菌和其他微生物,但在人体内并不起主要作用。嗜酸性粒细胞能合成多种细胞因子行使其功能,如:血小板活化因子,能促进创口愈合和纤维化的转化生长因子(TGF)α和β,与成人呼吸窘迫综合征和哮喘发病有关的巨噬细胞移动抑制因子(MIF),可能与Th2介导炎症反应有关的IL-12等。嗜酸性粒细胞也能生成和释放多种炎症介质。因此,嗜酸性粒细胞在参与正常免疫防御反应的同时,也能造成组织细胞的损伤。嗜酸性粒细胞中另一种主要的蛋白质成分(磷酸酯酶B)可形成夏-莱(Charcot-Leyden)结晶,见于与嗜酸性粒细胞增多有关疾病患者的痰、粪和组织内,常作为嗜酸性粒细胞相关疾病的标志。血中嗜酸性粒细胞计数并不总能反映组织受侵犯及损伤的程度。
【与嗜酸性粒细胞增多相关的疾病】
(一)寄生虫感染
是嗜酸性粒细胞增多最常见的原因。单细胞的原虫感染一般不引起嗜酸性粒细胞增高,而多细胞的蠕虫、吸虫感染可引起嗜酸性粒细胞增多,其增多的程度与虫体特别是幼虫侵入组织的数量和范围相平行。在组织内被包裹的或仅限于肠道腔内的感染(蛔虫、绦虫),一般不引起嗜酸性粒细胞增多。但能破坏肠黏膜的寄生虫(钩虫)可使嗜酸性粒细胞增多。临床上对原因不明的嗜酸性粒细胞增多者必须仔细了解其生活环境和饮食史,检查粪便以发现虫卵和幼虫。但有的寄生虫如旋毛虫、丝虫并不能从粪便中检出,因而,有寄生虫接触可能者、有哮喘发作、移位性肺炎、肝大等蚴虫移行征可疑者,必须进行有关的血液和组织学检查,以明确病因。
(二)变态反应性疾病
包括过敏性鼻炎、支气管哮喘、荨麻疹、血管神经性水肿和药物过敏反应等,均可出现嗜酸性粒细胞增多。药物过敏反应可仅表现为嗜酸性粒细胞增多,亦可引起间质性肾炎、血清病、胆汁淤积性黄疸、过敏性血管炎和免疫母细胞性淋巴结病等。一旦出现药物热和器官受累时应立即停药。药物引起的间质性肾炎嗜酸性粒细胞不但在血液内增多,而且在尿液中亦可检出。
(三)感染性疾病
某些传染病感染期,嗜酸性粒细胞常减少,恢复期可引起嗜酸性粒细胞增高,但猩红热急性期嗜酸性粒细胞常增高。有的真菌(曲菌、球孢子菌)感染和个别的慢性分枝杆菌病者,可有嗜酸性粒细胞增多。
(四)皮肤病
多种皮肤病如疥疮、天疱疮、疱疹性皮炎、剥脱性皮炎、湿疹、妊娠期疱疹和瘙痒性荨麻疹性丘疹及斑块综合征、银屑病、发作性血管神经性水肿等,均可伴嗜酸性粒细胞增多。
(五)肺嗜酸性粒细胞浸润症(pulmonary infiltration with eosinophilia,PIE)
是一组并不少见的疾病。其发病机制多与异常的免疫反应有关,但病因尚不确切。详见呼吸系统疾病见第十三篇第十章第八节。
(六)胃肠道疾病
嗜酸性粒细胞胃肠炎(eosinophilic gastroenteritis)的发病与变态反应有关,表现为消化不良、腹痛、腹泻和发热等。外周血白细胞分类中嗜酸性粒细胞可高达60%以上,消化道黏膜至浆膜层均可有嗜酸性粒细胞广泛浸润。病程可长达十余年,常呈自限性。此外,增多的嗜酸性粒细胞可存在于溃疡性结肠炎的病灶处。在溃疡性结肠炎和克罗恩病患者的血液中,有时也可有嗜酸性粒细胞增多。
(七)免疫性疾病
风湿性疾病(SLE、类风湿关节炎、结节性多动脉炎、皮肌炎等)、过敏性血管炎和肉芽肿性血管炎、部分先天性免疫缺陷、嗜酸性粒细胞筋膜炎药物治疗后及移植物抗宿主反应等,常有嗜酸性粒细胞增多。
(八)血液肿瘤
2008年WHO血液淋巴组织肿瘤分类新增一类疾病目录,名称为“髓系和淋系肿瘤伴嗜酸性粒细胞增多伴 PDGFRA , PDGFRB ,或 FGFR1 异常”。该目录包括三组罕见的疾病:①伴 PDGFRA 重排的髓系和淋巴系肿瘤;②伴 PDGFRB 重排髓系肿瘤;③伴 FGFR1 异常的髓系和淋巴系肿瘤(同义疾病名有8p11骨髓增生综合征、8p11干细胞综合征、8p11干细胞白血病/淋巴瘤综合征)。这三组疾病的发生机制均与编码异常酪氨酸激酶的融合基因形成有关。

(资源68) “疑难血液病临床和细胞形态学讨论”之八(病例)
已明确与 PDGFRA 或 FGFR1 相关的肿瘤起源于突变的多能(淋、髓系)干细胞,而与 PDGFRB 相关的肿瘤的起源是否也与多能(淋、髓系)干细胞突变相关尚不能完全确定。
所有三组疾病都可有慢性骨髓增生性肿瘤(MPN)的表现,但淋巴系肿瘤的发生率有差异。三组疾病不同的临床和血液学特征也与涉及的伴随基因有关。 PDGFRA 相关肿瘤通常表现为慢性嗜酸性粒细胞白血病(CEL)伴主要累及肥大细胞系列,有时也可累及中性粒细胞系列,而急性髓细胞白血病(AML)或前体T淋巴母细胞淋巴瘤(T-LBL)、以及两者共存伴嗜酸性粒细胞增多则较少见。 PDGFRB 相关肿瘤的MPN表现变化更大,常见于CMML伴嗜酸性粒细胞增多,异常的肥大细胞增生也可是其特征,向髓系急性变也有报道。 FGFR1 相关肿瘤以淋巴瘤表现常见,特别是伴嗜酸性粒细胞增多的T-LBL,也可见于CEL、前体B淋巴母细胞白血病/淋巴瘤以及AML。
(九)实体瘤
少数实体肿瘤,特别是能产生黏蛋白的上皮细胞来源的、转移至浆膜及骨骼的、病灶中心有坏死的癌肿和肉瘤患者,血液中亦可见嗜酸性粒细胞增多。朗格汉斯细胞组织细胞增生症骨嗜酸肉芽肿多见于婴幼儿和青少年,病变主要侵犯骨组织。病理示大量组织细胞和嗜酸性粒细胞浸润,但外周血嗜酸性粒细胞多<10%。Kimura病(Kimura disease)常以头颈部肉芽肿病变伴外周血嗜酸性粒细胞增高为主要临床表现。Kimura病是一种病因不明的嗜酸性粒细胞增生性淋巴肉芽肿。
(十)异常表型的克隆性T淋巴细胞
2004年以来,国外不少学者证明部分特发性高嗜酸性粒细胞综合征患者体内存在着异常克隆的T淋巴细胞亚群,这些异常的克隆性T淋巴细胞的表型以CD3 - CD4 + CD8 - 为多见。由于异常克隆T淋巴细胞产生大量的IL-2、IL-3、IL-5以及GMCSF,因此可导致酸粒细胞大量增生。现认为存在异常表型克隆T淋巴细胞的特发性高嗜酸性粒细胞综合征患者有可能发展成为T细胞淋巴瘤。
(十一)慢性嗜酸性粒细胞白血病,非特指(chronic eosinophilic leukemia,not otherwise specified,CEL,NOS)和特发性高嗜酸性粒细胞综合征(idiopathic hypereosinophilic syndrome,特发性HES)
CEL,NOS和特发性HES究竟同属一组异质性的疾病还是为两种不同疾病实体,目前还未完全阐明。WHO(2001)髓系肿瘤分类将两者以CEL/HES的病名列入慢性骨髓增生性疾病之中,并特别说明若要诊断CEL,就应该有嗜酸性粒细胞克隆性增生或骨髓原始细胞增多的证据,如无这方面的证据,就宁可诊断特发性HES。CEL/HES在临床上较为罕见,美国2001—2005年的流行病学资料显示,CEL/HES年龄调整后的年发病率为0.036/10万,男性约为女性的1.5倍。修订后的 WHO(2008)分类将CEL,NOS确定为骨髓增殖性肿瘤(MPN)中的一个疾病实体,并且强调诊断CEL,NOS必须确定不存在 PDGFRA , PDGFRB ,或 FGFR1 异常。而特发性 HES应在CEL,NOS诊断条件不能满足前提下作出。此外,还提出了特发性嗜酸性粒细胞增多(idiopathic hypereosinophilia)的诊断建议。
CEL,NOS与特发性HES的临床表现甚难区分。两者均可因嗜酸性粒细胞浸润组织并释放细胞因子和体液因子,导致全身多个器官的损伤。心、肺、中枢神经系统、皮肤和胃肠道最常受累,严重者出现心肌内膜纤维化、限制性心肌肥大,心脏瓣膜瘢痕导致瓣膜性回流和附壁血栓形成,可引起脑栓塞、周围神经病变等。30%~50%的患者有肝、脾受累。还常见风湿病样表现。但也有10%左右的患者并无明显症状,偶然因查血常规发现嗜酸性粒细胞增多。
实验室检查外周血最显著的特点是嗜酸性粒细胞增多,通常主要为成熟嗜酸性粒细胞,可有不同程度的嗜酸性粒细胞形态异常。但因这些变化也可见于反应性嗜酸性粒细胞增多患者,因此对诊断无多大帮助。骨髓检查增生极度活跃,多数以成熟嗜酸性粒细胞为主,部分患者可有骨髓纤维化。应重视检查外周血和骨髓的原始细胞数量,如外周血>2%或骨髓>5%,应倾向于诊断CEL,NOS。
(十二)其他
浆膜表面受刺激,如炎症、腹部照射、长期腹膜透析、损伤或反复穿刺等,可引起浆膜腔积液及血液中嗜酸性粒细胞增多。严重的中毒性疾病、嗜酸性粒细胞增多肌痛综合征、肾上腺及垂体功能低下者,可引起嗜酸性粒细胞增多。
【诊断】
外周血中嗜酸性粒细胞绝对值增高即可诊断为嗜酸性粒细胞增多症。关键是明确病因诊断。最常见为反应性嗜酸性粒细胞增多症和继发性嗜酸性粒细胞增多症,见于寄生虫感染、过敏性疾病、肺嗜酸性粒细胞浸润症、皮肤病、结缔组织病、Kimura病和肿瘤等。必须详尽地询问病史和进行全面的体格检查,选用必要的辅助诊断方法加以明确。特别须注意有否造血系统肿瘤包括髓系肿瘤和淋系肿瘤伴嗜酸性粒细胞增多。有条件单位应常规作骨髓细胞染色体核型分析和涉及 PDGFRA , PDGFRB ,或 FGFR1 融合基因、T细胞受体基因重排等分子遗传学检测,以确定是否为克隆性嗜酸性粒细胞增多症。对诊断一时不能肯定者应密切随访。
WHO(2008)分类提出的CEL,NOS诊断标准为:①外周血嗜酸性粒细胞增多≥1.5×10 9 /L;②无Ph染色体或 BCR-ABL1 融合基因,无其他 MPN或 MDS/MPN;③无t(5;12)(q31-35;p13)或其他 PDGFRB 重排;④无 FIP1 L1-PDGFRA 融合基因或其他 PDGFRA 重排;⑤无 FGFR1 重排;⑥外周血原始细胞和骨髓原始细胞<20%,无inv(16)(p13;q22)或t(16;16)(p13;q22),无 AML其他特征;⑦有克隆性细胞遗传学或分子遗传学异常,或外周血原始细胞>2%,或骨髓原始细胞>5%。特发性HES的诊断标准须符合:①外周血嗜酸性粒细胞增多≥1.5×10 9 /L至少6个月;②除外反应性和继发性嗜酸性粒细胞增多症;③除外髓系肿瘤包括AML、MPN、MDS、MDS/MPN和系统性肥大细胞增生症;④除外具有免疫表型异常,细胞因子产生异常的T细胞群的疾病;⑤具有因嗜酸性粒细胞增多产生的组织损害。若只符合上述标准中的①~④,则应该诊断为特发性嗜酸性粒细胞增多,以便与特发性HES相鉴别。
【治疗】
反应性和继发性嗜酸性粒细胞增多的处理以治疗原发病为首要。非克隆性嗜酸性粒细胞增多治疗目的在于抑制嗜酸性粒细胞的生成。初用泼尼松1mg/(kg·d)可使约1/3的患者得到缓解,用药一般需持续2个月,见效后逐渐减量至能控制疾病的最小剂量。疗效不佳者可加用羟基脲口服。剂量为0.5~1.5g/d,维持白细胞计数在(4~10)×10 9 /L。如嗜酸性粒细胞计数大于100×10 9 /L,应考虑白细胞单采术。α干扰素对改善心功能和心肌损害有效。有血栓并发症者可用抗血小板药物或华法林抗凝治疗。心脏瓣膜受累者偶需外科手术。
对于有 FIP1 L1-PDGFRA 融合基因阳性和 PDGFRB 重排者,近年来取得的重大进展是酪氨酸激酶抑制剂伊马替尼疗效瞩目。国外一项多中心前瞻性Ⅱ期临床试验的研究结果显示, FIP1 L1-PDGFRA 融合基因阳性的患者口服伊马替尼每日从100mg逐步增至400mg,可使所有患者取得完全血液学缓解及融合基因转录本转为阴性,但需要维持给药。对于部分 FIP1 L1-PDGFRA 融合基因阴性的CEL/HES患者,伊马替尼也有一定的疗效,据推测这些患者可能存在着未知的隐蔽基因异常。但 FGFR1 相关肿瘤对现有的酪氨酸激酶抑制剂治疗无效。此外,IL-5单克隆抗体、CD52单克隆抗体及自体造血干细胞移植也正在试用于临床治疗,其确切疗效有待于进一步观察。
主要参考文献
1.张之南,杨天楹,郝玉书,等.血液病学.第2版.北京:人民卫生出版社,2012,562-582.
2.Swerdlow SH,Campo E,Harris NL et al.(eds).WHO Classification of Tumors of Haematopoietic and Lymphoid Tissues.4th ed.Lyon:IARC,2008,31-86.
3.Gotlib J,Cools J,Malone JM,et al.The FIP1L1-PDGFRα fusion tyrosine kinase in hypereosinophilic syndrome and chronic eosinophilic leukemia:implications for diagnosis,classification,and management.Blood,2004,103(8):2879-2891.
4.Cools J.The hypereosinophilic syndrome:idiopathic or not,that is the question.Haematologica,2005,90(5):582-584.
5.Tefferi A and Vardiman JW.Classification and diagnosis of myeloproliferative neoplasms:The 2008 world health organization criteria and point-of-care diagnostic algorithms.Leukemia,2008,22:14-22.
6.Baccarani M,Cilloni D,Rondoni M,et al.The efficacy of matinib mesylate in patients with FIP1L1-PDGFRa-positive hy-pereosinophilic syndrome.Results of a multicenter prospective study.Haematologica,2007,92(9):1173-1179.
第六节
系统性肥大细胞增多症
许小平
肥大细胞增生症(mastocytosis)是由于克隆性、肿瘤性增生的肥大细胞积聚在一个或多个器官的一组异质性疾病。由于肥大细胞来源于造血系统的前体细胞,因此肥大细胞增生症也属造血系统疾病。2008年WHO髓系肿瘤分类标准将其列入骨髓增殖性肿瘤(myeloproliferative neuplasms,MPN)之中。肥大细胞增生症的临床表现呈多样化,从自限性的皮肤病变到累及全身多器官的高度侵袭性肿瘤不等。肥大细胞增生症归分为7类:①皮肤肥大细胞增生症(CM);②惰性系统性肥大细胞增生症(indolent systemic mastocytosis,ISM);③系统性肥大细胞增生症伴非肥大细胞系克隆性血液增生疾病(systemic mastocytosis with associated clonal hematological non-mast cell lineage disease,SM-AHNMD);④侵袭性系统性肥大细胞增生症(aggressive systemic mastocytosis,ASM);⑤肥大细胞白血病(mast cell leukemia,MCL);⑥肥大细胞肉瘤(mast cell sarcoma,MCS);⑦皮肤外肥大细胞肿瘤(extracutaneous mastocytoma)。皮肤肥大细胞增生症是指肥大细胞的增生局限于皮肤,包括色素性荨麻疹/斑丘疹、弥漫性皮肤肥大细胞增生症及孤立性皮肤肥大细胞瘤3类。本节着重介绍属系统性肥大细胞增生症(systemic mastocytosis,SM)的4类,即ISM、SM-AHNMD、ASM 及 MCL。
【流行病学】
基于系统性肥大细胞增生症在临床上少见,临床医生对本病的诊断有一个逐步熟悉的过程,所以可靠的流行病学调查文献甚少。最近丹麦的548例成人SM的资料研究显示发病率为0.89/10万。男性发病多见,占59.9%。发病平均年龄为49.6岁,最常见的亚型是SM,占82%,其次为亚型不明占11%,SM-AHNMD占4%,SM为2%,MCL仅1%。但MCL诊断时年龄为75.4岁,男性占80%。罕有家族性病例报道。
【临床特点】
系统性肥大细胞增生症在诊断时的表现和症状取决于SM的亚型、肥大细胞释放的介质和器官受浸润程度。可归纳为以下几类:①体质性症状:如乏力、体重减轻、发热和出汗;②皮肤表现:可有瘙痒、风疹和皮肤地图样变等;③介质相关症状:常见有腹痛、胃肠道不适、脸红、昏厥、高血压、头痛、低血压、心动过速、呼吸道症状等;④骨相关症状:包括骨痛、骨折和关节痛等。体检可发现脾脏肿大,而淋巴结肿大和肝脏肿大相对少见。
【实验室检查】
多数患者有血液学的异常。贫血、白细胞与血小板增多或减少都可能出现,有的患者可见到嗜酸性粒细胞增多。除个别肥大细胞白血病的患者外,血常规中肥大细胞不常见。
骨髓活检多数患者在骨小梁旁和血管周围可见到多灶性、界限清楚的肥大细胞聚集。通常病灶的中心是淋巴细胞,周围有肥大细胞包绕,边缘是反应性嗜酸性粒细胞。部分患者病变呈单一的形态特征,主要由梭形肥大细胞沿骨小梁边缘呈串状分布。其他常被累及的器官有脾、淋巴结、肝和胃肠道。50%以上的患者可出现皮肤病变。
正常肥大细胞缺乏髓过氧化物酶,但存在萘酚-ASD-氯乙酸酯酶。CD45、CD33、CD68和CD117表达阳性,CD14、CD15、CD16及T细胞和B细胞相关抗原表达阴性。所有肥大细胞表达类胰蛋白酶,而糜蛋白酶仅表达于部分亚群。肿瘤性的肥大细胞除表达类似正常肥大细胞的抗原外,还可表达CD2和CD25。
类胰蛋白酶是特异性存在于肥大细胞中的中性蛋白酶,占肥大细胞中蛋白酶含量的25%。血浆总类胰蛋白酶水平反映肥大细胞的载量,因此可用于肥大细胞增生症的诊断及病情评估,尤其在与IgE升高的其他过敏性疾病相鉴别时有重要价值。血浆总类胰蛋白酶正常值1~15ng/ml,平均5ng/ml。系统性肥大细胞增生症患者,常高于>20ng/ml,甚至可超过200ng/ml。但应注意当机体发生超敏反应或存在其他肿瘤性骨髓增生异常时血浆类胰蛋白酶也可一过性升高。
【诊断】
(一)WHO(2008)诊断标准
1.主要标准
在骨髓的组织切片中或其他非皮肤器官中可见多灶性密集的肥大细胞浸润(≥15个肥大细胞聚集)。
2.次要标准
(1)在骨髓活检切片或其他非皮肤器官中,>25%浸润的肥大细胞为梭型细胞样或非典型形态;或骨髓穿刺涂片所有肥大细胞中>25%的为幼稚或非典型。
(2)骨髓、血液或其他非皮肤器官可检出位于密码子816的 KIT 基因点突变。
(3)骨髓、血液或其他非皮肤器官的肥大细胞除了正常肥大细胞标记外,同时表达CD2和(或)CD25。
(4)血浆总类胰蛋白酶浓度持续>20ng/ml(如伴有髓系克隆性异常疾病,此参数无效)。
具备1个主要标准和1个次要标准,或3个次要标准即可诊断系统性肥大细胞增生症。
(二)SM各亚型诊断标准
WHO(2008)分类标准将与SM相关的临床特征分为B症状和C症状。B症状为:①骨髓活检显示肥大细胞浸润>30%(灶性,致密聚集),和(或)血清总胰蛋白酶>200ng/ml;②骨髓非肥大细胞系的异常增生和发育异常,但不足以诊断为WHO分类中的任一种造血组织肿瘤,外周血细胞计数正常或轻度异常;③肝大但不伴肝功能损害,和(或)脾大但不伴功能亢进,和(或)浅表或深部淋巴结肿大。C症状为:①骨髓功能异常,表现为一系或多系血细胞减少(ANC<1×10 9 /L,Hb<100g/L,血小板<100×10 9 /L),但没有非肥大细胞恶性肿瘤的证据;②肝大伴肝功能损伤,腹水,和(或)门脉高压;③骨累及伴有大面积的溶骨性损害和(或)病理性骨折;④脾大伴脾功能亢进;⑤胃肠道肥大细胞浸润致营养吸收不良伴体重下降。
1.惰性系统性肥大细胞增生症(ISM)
符合SM的诊断标准;无C症状;无非肥大细胞系克隆性造血组织恶性肿瘤/疾病的证据。骨髓肥大细胞增生症(bone marrow mastocytosis)指符合ISM,伴有骨髓累及,但无皮肤病变;冒烟型系统性肥大细胞增生症(smouldering systemic mastocytosis)指符合ISM,但有2项以上B症状,无C症状。
2.系统性肥大细胞增生症伴非肥大细胞系克隆性血液增生疾病(SM-AHNMD)
指同时符合SM标准和其他相关克隆性血液非肥大细胞疾病(MDS,MPN,AML,淋巴瘤,或其他符合WHO分类的淋巴造血肿瘤)标准。
3.侵袭性系统性肥大细胞增生症(ASM)
符合SM的标准;1项或更多项C症状;无肥大细胞白血病的证据。伴嗜酸性粒细胞增多的淋巴结病性肥大细胞增生症(lymphadenopathic mastocytosis with eosinophilia)指具有侵袭性淋巴结病,伴外周血中嗜酸性粒细胞增多,常伴广泛骨累及和肝脾大,一般无皮肤病损的的病例,但须除外PDGFRA重排阳性患者。
4.肥大细胞白血病(MCL)
符合SM的诊断标准;骨髓活检见不典型的不成熟肥大细胞弥漫性间质浸润;骨髓涂片见≥20%的肥大细胞;典型病例外周血白细胞中肥大细胞≥10%;如外周血白细胞中肥大细胞<10%,可诊断为非白血性肥大细胞白血病(aleukemic mast cell leukemia)变异型。
【治疗】
系统性肥大细胞增多症的治疗强调个体化,其个体化的主要依据是患者的亚型分类。如ISM可能并不影响预期寿命,治疗措施应以对症处理为主,包括避免肥大细胞炎性介质释放的激发、控制炎性介质的释放以及治疗肥大细胞对的器官浸润。
激发肥大细胞炎性介质释放的常见因素有温度改变、劳累、情绪焦虑;某些药物如阿司匹林、非类固醇类消炎药、阿片类止痛剂、全身麻醉用的肌肉松弛剂等;此外,酒精、蛇毒与昆虫叮咬也可激发炎性介质的释放。
组胺H 1 和H 2 受体拮抗剂对组胺引起的相关症状有效。此外还可选用糖皮质激素、色甘酸钠及抗胆碱能药等。如出现过敏性休克,给予肾上腺素治疗。
由于阿司匹林可促使肥大细胞释放介质,导致血管性虚脱,一般不作为常规使用。麻醉药物能激活肥大细胞释放血管活性介质,产生明显的全身症状甚至死亡。因此患者应用麻醉剂时必须联合应用组胺H 1 和H 2 受体拮抗剂。
有肿瘤细胞显著增生导致器官浸润者可考虑应用骨髓抑制性药物单药或联合治疗,以减少器官浸润程度。干扰素-α 2b 第1周剂量为300万U/次,每周3次,以后根据疗效和副作用调整剂量。在使用干扰素-α 2b 前几天可给予泼尼松每日50~75mg口服。也可应用2-氯脱氧腺苷5mg/(m 2 ·d)×5天,每4~6周为1个周期。有 C-KIT 突变者可试用伊马替尼,据报道对伴有嗜酸性粒细胞增多者有效;如效果不理想也可试用达沙替尼。
目前尝试按照异常的分子遗传学特征开展治疗的空间还极其局限,现有的 KITD816 V 抑制剂基本无效,可能与SM发病的分子信号调控通路尚未完全认识有关。但有个案报道伊马替尼对转膜 KIT 突变(如F522C或K509I)的SM患者有效。最近正在研究中的一些新抑制剂(例如midostaurin/PKC412)是否能够真正开启SM的分子靶向治疗还有待评估。异基因造血干细胞移植治疗SM的经验有限。最近报道57例(中位年龄46岁)接受异基因造血干细胞移植的SM患者,其中38例为SM-A HNMD(20例伴AML)、12例 MCL、7例ASM。移植后100天,16例(28%)获得完全缓解,其中2例 KITD816 V 转阴性;24例(42%)获得部分缓解;12例(21%)疾病稳定。原发性难治主要见于 MCL患者。伴随的造血系统恶性肿瘤对治疗更为敏感。3年整体存活(OS)55%。其中SM-AHNMD亚型为74%。OS危险因素:MCL(与ASM或SM-AHNMD比较)、减低剂量预处理(与清髓预处理比较)、疾病进展(与有治疗反应或疾病稳定比较)。
【预后】
预后取决于患者所属疾病的种类。侵袭性者如肥大细胞白血病,仅能存活几周至数月。而惰性的系统性肥大细胞增生症通常不影响预期寿命。SM影响预后的一个重要指标是有无皮肤的累及。无皮肤累及常为侵袭性病变,反之,则多呈惰性过程。
主要参考文献
1.Arock M,Akin C,Hermine O,et al.Current treatment op-tions in patients with mastocytosis:status in 2015 and future perspectives.Eur J Haematol,2015,94(6):474-490.
2.Pardanani A.Systemic mastocytosis in adults:update on di-agnosis,risk stratification,and management.Am J Hematol,2015,90(3):250-262.
3.Tefferi A,Verstovsek S,and Pardanani A.How we diagnose and treat WHO-defined systemic mastocytosis in adults.Haematologica,2008,93(1):6-9.
第七节
慢性中性粒细胞白血病
许小平
慢性中性粒细胞白血病(chronic neutrophilic leukemia,CNL)属MPN的少见类型。1920年Tuohey首先报道本病。WHO(2001)造血与淋巴肿瘤诊断分类将其作为一个独立的疾病实体列出,当时文献报道不足150例。以后按WHO诊断标准又陆续报道过40例。CNL中位发病年龄为67岁(范围26~80岁)。其中男性占57%,36%患者以前有细胞毒暴露史。
2013年CNL的发病机制研究获得重大进展。Maxson等发现本病是一种CSF3R(colony-stimulating factor 3 receptor,集落刺激因子3受体)膜近端区域突变所致的疾病。集落刺激因子3(CSF3)是中性粒细胞产生的主要刺激因子,CSF3R膜近端区域突变可影响中性粒细胞的生成和存活信号通路。CNL最多见的突变类型是膜近端区域CSF3RT618I,另外还可有CSF3RM696T和CSF3RI598I。
白细胞计数增高是本病的突出表现,中位值为39×10 9 /L,最高可达126×10 9 /L增高以中性粒细胞为主。外周血涂片中性粒细胞通常为分叶核,但杆状核也可明显增多。绝大多数患者未成熟阶段的粒细胞(早幼粒细胞、中幼粒细胞、晚幼粒细胞)<5%,偶尔可达10%。但不能找到原始粒细胞。中性粒细胞可见异常粗大的中毒颗粒,也可以是正常形态。红细胞和血小板无形态异常。骨髓检查示增生极度活跃,中性粒细胞增多,粒红比例高达20∶1甚至以上。原始粒细胞和早幼粒细胞不增多,以中幼粒细胞和成熟粒细胞增多为主。可以有红系和巨核系增多,各系均无明显发育异常。网状纤维一般不增多。
患者常有轻度贫血,血红蛋白中位值大约为110g/L。血小板计数正常或轻度下降,但晚期常呈下降趋势,尤其在脾脏逐渐增大的情况下。中性粒细胞碱性磷酸酶积分增高。血清维生素B 12 明显升高,血清尿酸和乳酸脱氢酶也可升高。几乎90%的患者染色体检查为正常。其余的核型异常有+8、+9、del(20)和 del(11q),无 Ph染色体或 BCR / ABL 融合基因。
羟基脲、干扰素-α和阿糖胞苷对于降低白细胞数有效,也可使肿大的脾脏缩小,但很少能获得长期疗效。不推荐蒽环类联合阿糖胞苷的强烈化疗。Ruxolitinib(JAKs抑制剂)可能有一定疗效。条件许可患者可考虑行异基因造血干细胞移植。

