第五章
淋巴细胞与浆细胞疾病
第一节
淋巴瘤概述

陈波斌
淋巴瘤(lymphoma)是一组异质性的肿瘤性疾病,起源于发生突变的单个淋巴细胞,突变后的淋巴细胞具有增殖和生存优势。淋巴瘤可发生于身体的任何部位,淋巴结、扁桃体、脾及骨髓最易受到累及。无痛性、进行性淋巴结肿大和局部肿块是其特征性的临床表现,可伴有某些器官的受压迫症状。病变侵犯结外组织如胃肠道、骨骼或皮肤等,则表现为相应组织器官受损的症状,当淋巴瘤浸润骨髓时可形成淋巴瘤细胞白血病。常有发热、消瘦、盗汗等全身症状。以往认为淋巴瘤与淋巴细胞白血病是两种疾病,但近年来随着基础研究的不断深入,人们发现这两类肿瘤的疾病本质难以明确区别。根据组织病理学特征将淋巴瘤分为霍奇金淋巴瘤(Hodgkin lymphoma,HL)和非霍奇金淋巴瘤 (non-Hodgkin lymphoma,NHL)两大类,85%的淋巴瘤为NHL。
【淋巴瘤分类的演变】
淋巴瘤是一组来源于B细胞、T细胞或NK细胞的异质性的肿瘤。早在19世纪末20世纪初,病理学家就命名并定义了霍奇金淋巴瘤、淋巴肉瘤等疾病名称。1942年,Gall和Mallory提出分为淋巴母细胞淋巴瘤和淋巴细胞性淋巴瘤两类;网状细胞肉瘤分为干细胞淋巴瘤和破折细胞性淋巴瘤;还纳入了滤泡型淋巴瘤这一新的病种。这个分类被认为是第一个真正意义上的淋巴瘤分类。1966年Rappaport分类直接将“分化”的概念引入分类中,分为“未分化”、“分化差”、“分化好”等类型;所有类型的淋巴瘤(包括HL和NHL,淋巴细胞性和组织细胞性),其生长方式都可分为结节型和弥漫型两类,并认为结节型预后比弥漫型为好。作者还用组织细胞性淋巴瘤取代网状细胞肉瘤,未分化淋巴瘤取代干细胞淋巴瘤。20世纪70年代,还有BNLL分类(1974)、Kiel分类(1974)、Rappaport修改分类(1974)、Dorfman分类(1974)、Lukes-Collins修改分类(1974)以及WHO分类(1976)等多个分类,但这些诊断分类系统没有一种被广泛认可与应用。
1982年,美国国立癌症研究所(NCI)的工作分类(working formulation)将淋巴瘤分为高度恶性、中度恶性和低度恶性三类,因简单实用,被广泛应用于临床实践。我国学者结合中国淋巴瘤的特点,1985年制订了成都会议分类。
随着对于免疫系统和淋巴细胞发育规律的认识不断深入,针对不同类型淋巴细胞的单克隆抗体和淋巴细胞基因表达谱不断地被开发和应用,1992年和1994年又对Kiel分类和Lukes-Collins分类进行修改,将非霍奇金淋巴瘤明确区分为B细胞和T细胞两大类。
1994年国际淋巴瘤研究组提出了修订的欧美淋巴瘤分类标准(REAL分类),将淋巴瘤的诊断建立在病理组织学、免疫学和细胞遗传学的基础上。这个分类可以认为是第一个真正具备现代科学意义的淋巴瘤分类、在REAL分类基础上,由欧洲血液病理学家学会和美国血液病理学会发起,共有125位世界各国病理学家和临床医生参与制订的WHO关于造血与淋巴组织肿瘤的分类标准在2001年问世。2008年又根据最新研究进展进行了修订更新。WHO(2008)淋巴组织肿瘤分类标准主要有如下几个重要特点:
1.淋巴瘤分为B细胞、T/NK细胞、霍奇金淋巴瘤三大类。
2.根据肿瘤细胞的分化阶段将B细胞肿瘤、T细胞和NK细胞肿瘤分为前体细胞肿瘤和成熟细胞肿瘤两类。
3.强调对疾病本质的认识,尤其重视免疫学、细胞遗传学和分子生物学检测结果在诊断分类中的价值,不再将淋巴瘤和淋巴细胞白血病视为两类不同性质的疾病实体。
4.将浆细胞肿瘤(如多发性骨髓瘤、华氏巨球蛋白血症等)也归属于成熟B淋巴细胞肿瘤之内。
5.首次提出“灰区”淋巴瘤的概念。
【流行病学】
淋巴瘤的发病男性多于女性,随年龄的增加而增加。不同国家淋巴瘤的发病率存在差异。我国淋巴瘤的发病率明显低于欧美各国及日本。据全国肿瘤登记中心报告,2012年我国淋巴瘤的发病率为6.13/10万,年龄标化发病率为4.81/10万,在男性位于肿瘤第10位。淋巴瘤可发生于任何年龄,最小为3个月,最大>80岁,以20~40岁为多见,约占50%;城市的发病率高于农村。我国2012年淋巴瘤死亡率为3.18/10万,年龄标化死亡率为2.3/10万,占男性恶性肿瘤死亡的第9位。淋巴瘤发病率有逐年上升的趋势,1950—1990年全世界NHL的死亡率增加了1.5倍,可能与环境污染、寿命延长以及诊断水平的不断提高等有关。不同区域淋巴瘤的亚型分布也存在差异,在美国滤泡型淋巴瘤约占NHL的30%,但在许多发展中国家包括中国相对较少,Burkitt淋巴瘤最常见于赤道非洲国家,T细胞白血病/淋巴瘤最常见于日本西南部、美国东南部、南美东北部和加勒比盆地。
虽然大多数淋巴瘤无明确病因,但人类T细胞白血病/淋巴瘤病毒Ⅰ型、EB病毒、丙肝病毒、人类疱疹病毒-8感染及幽门螺杆菌和衣原体与淋巴瘤的发生有很强的相关性。一些职业或工业暴露可能与淋巴瘤的发生有一定关系,如有机氯、杀虫剂等。从遗传易感性上看,淋巴瘤或其他血液肿瘤的患者的同胞淋巴瘤的发病风险增加。
【预后】
淋巴瘤是一类高度异质性的血液肿瘤疾病,临床过程、对治疗的反应和预后不仅与不同的病理学类型有关,甚至同一种病理学类型的不同患者也存在差异。既往建立在患者临床特征为基础的疾病预后判断指标(如IPI、FLIPI等)目前仍广泛地应用于临床,越来越多的免疫学和分子生物学检测指标可能从本质上更好地预测疾病的结果,为临床治疗决策提供客观依据。
主要参考文献
1.Swerdlow SH,Campo E,Harris NL,et al.WHO Classification of Tumors of Haematopoietic and Lymphoid Tissues.4th ed.Lyon:IARC,2008,179-350.
2.陈万青,郑容寿,张思维,等.2012年中国恶性肿瘤发表和死亡分析.中国肿瘤,2016,25(1):1-8.
第二节
霍奇金淋巴瘤
陈波斌
霍奇金淋巴瘤(Hodgkin lymphoma,HL),是一种B细胞淋巴瘤,大多起源于生发中心,主要累及淋巴结、脾、肝脏和骨髓。HL的发病率以北美、北欧最高,东亚的发病率较低。在我国HL占淋巴瘤的8%~11%,与国外占25%有显著不同。男、女性别比为1.2∶1,中位发病年龄38岁,发病年龄有两个高峰:15~34岁和60岁以上。10岁以下者罕见。
【病因】
(一)病毒感染
曾患传染性单核细胞增多症的EB病毒感染者发生HL的风险增加了3倍,HL患者血清EB病毒衣壳抗体的滴度显著高于对照组,而且在发生肿瘤以前已存在数年。原位杂交研究显示约50%HL患者的Reed-Sternberg细胞(R-S细胞)内可以检出含有EB病毒编码的小RNA,这种情况尤其易见于混合细胞亚型及年龄大于60岁的患者。HIV感染与霍奇金淋巴瘤的发病也有密切关系,HIV感染者发生霍奇金淋巴瘤的风险是正常人群的10~20倍。
(二)遗传因素
遗传易感性在霍奇金淋巴瘤的发病中也发挥了重要作用,有文献报道4.5%的患者家族中有人患霍奇金淋巴瘤或其他肿瘤;单卵双生同胞之一发生HL,另一同胞发生该病的风险是异卵双生者的100倍。HL患者第一代亲属发生该病的风险增加5倍。具有自身免疫性疾病尤其结节病病史或家族史者,霍奇金淋巴瘤的患病风险增加。
(三)其他免疫性疾病
移植后应用免疫抑制剂的患者、先天性免疫缺陷者(如共济失调毛细管扩张、Klinefelter综合征、Chediak-Higashi综合征、Wiskott-Aldrich 综合征)及自身免疫性疾病患者(类风湿关节炎、非热带性口炎性腹泻,Sjögren综合征,系统性红斑狼疮等)可轻度增加HL的发病风险。
【临床表现】
(一)淋巴结肿大
大多数为无痛性颈部或锁骨上淋巴结进行性肿大(占60%~70%),其次(约30%)为腋下淋巴结肿大,横膈下淋巴结肿大约为10%左右。肿大的淋巴结可以活动,也可互相粘连,融合成块,触诊有软骨样感觉。少数患者仅有深部淋巴结肿大,表现为纵隔或后腹膜肿块。HL侵犯各器官可引起相应部位的病变及症状,骨髓受累(发生率低于10%)常无症状。颅内、胃和皮肤损害罕见。
(二)发热及其他特殊表现
25%的患者出现全身症状,部分患者以原因不明的持续发热为首发表现,通常年龄稍大,男性多见,常有腹膜后淋巴结累及。约1/6的患者出现周期性发热(Pel Ebstein热),表现为有规律的高热数天后体温恢复至正常或低于正常,维持数天后再次发热。当疾病累及单核-吞噬细胞系统以外,可表现为局部或全身皮肤瘙痒,多见于年轻患者,尤其女性,瘙痒甚至是HL的唯一全身症状,在确诊前数年即已出现。部分患者在饮用含酒精饮料后在病变部位可出现疼痛,机制不明。随着疾病进展,恶病质常见。
部分霍奇金淋巴瘤患者在确诊时存在肿瘤伴发性综合征表现,包括胆管消失综合征和特发性胆管炎、肾病综合征、自身免疫性血液病(如免疫性血小板减少或溶血性贫血)等。霍奇金淋巴瘤神经系统的肿瘤伴发性综合征包括亚急性小脑变性、脊髓病进行性多灶性脑病和边缘叶脑炎。
【实验室检查与特殊检查】
(一)血液和骨髓检查
常有轻到中度贫血,通常发生于疾病晚期,贫血常为慢性病贫血,很少发生溶血性贫血;白细胞可轻度或明显增加,以中性粒细胞增多为主,约1/5的患者嗜酸性粒细胞升高;可见血小板增多。血小板减少的病因除脾功能亢进外,也可由免疫因素或骨髓受累等所致。血细胞减少尤易见于疾病处于进展期及淋巴细胞削减型霍奇金淋巴瘤。
如果患者属高危人群或病变部位特殊,需做HIV抗体检查。对于就诊时有B症状(发热、盗汗和体重减轻)或外周血细胞计数低于正常水平的患者,应做骨髓活检和涂片检查。12%的初诊患者有骨髓受累,尤其见于老年、进展期、组织学预后不良、有全身症状或免疫缺陷者。年轻、无症状且临床分期为Ⅰ或Ⅱ期者,很少出现骨髓受累及。骨髓涂片找到R-S细胞是HL浸润骨髓的依据;骨髓穿刺涂片阳性率仅3%,但骨髓活检可提高至9%~22%。疾病活动期血沉增快,血清乳酸脱氢酶活性增高,后者提示预后不良。血清碱性磷酸酶增高,但并非一定表明骨髓或肝脏受累。
(二)影像学检查
所有患者均应做胸部、腹部和盆腔的CT检查,当骨骼或软组织受累及或静脉应用造影剂有禁忌时进行磁共振检查。PET-CT用于疾病的分期检查和治疗后残留病灶的检查灵敏度和特异度高于CT或 67 镓扫描。PET-CT最大优点是能够更准确、全面地进行疾病的分期,但需要注意的是PET-CT扫描判断骨髓是否存在病变时可出现假阳性,这在化疗后骨髓造血恢复或应用造血细胞集落刺激因子时易于出现。在随访过程中,当出现胸腺增生、肉芽肿病或感染性疾病时也可导致PET-CT出现假阳性。尤其是此前CT未发现的病灶,需要组织活检以进一步确诊。
(三)病理活组织检查
病理学检查是确诊HL的基本方法。选取较大的淋巴结,完整地取出,避免挤压。深部淋巴结可在B超或CT引导下细针穿刺涂片做细胞病理形态学检查,但单纯细针穿刺往往不能确诊HL;如果只有纵隔淋巴结肿大,最好采用纵隔镜对纵隔淋巴结进行活检;也可以考虑CT引导活检。
(四)剖腹探查
如发热待查病例,临床高度怀疑淋巴瘤,B超发现有腹腔淋巴结肿大,但无浅表淋巴结或病灶可供活检的情况下,宜选择剖腹探查或腹腔镜活检,尤其后者对患者的创伤较小,值得推荐。但单纯为了临床分期,不推荐剖腹探查。
【病理与诊断】
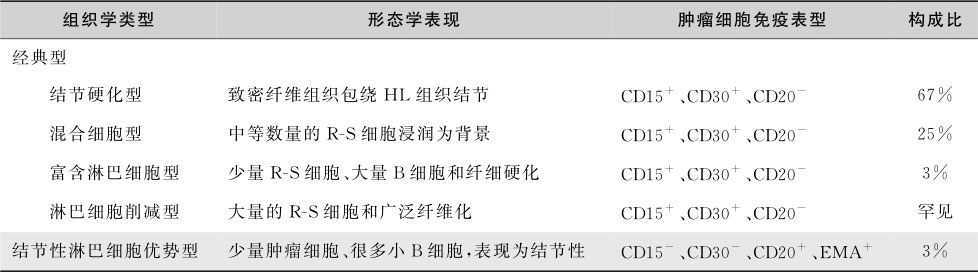
H L确诊依赖于病理检查,在淋巴结或结外组织如骨髓、肺或骨骼组织中,可找到R-S细胞。WHO(2008)分类中,将HL分为经典型和结节性淋巴细胞优势型(表16-5-1),前者又分为结节硬化型、混合细胞型、富含淋巴细胞型和淋巴细胞削减型。组织分型与预后有密切关系。经典型霍奇金淋巴瘤中以结节硬化型最常见,混合细胞型次之,其他各型均较少见。免疫组织化学检查为HL的分型诊断提供了依据:经典型HL的R-S细胞免疫表型为 :CD30 + (80%~100%病例 )、CD15 + (75%~85%病例)、B细胞特异性的激活蛋白(BSAP)(+)(>90%的病例),但只有少数恶性细胞CD15和BSAP染色阳性。约有40%的经典型CD20弱阳性;几乎所有结节性淋巴细胞优势型CD20、CD79a和CD45强阳性,CD30和CD15阴性。
表16-5-1 不同亚型霍奇金淋巴瘤的病理学特点和构成情况
【鉴别诊断】
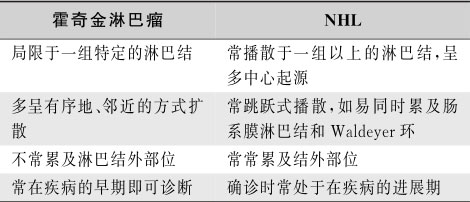
以发热为主要表现的HL,须和结核病、败血症、结缔组织病、坏死性淋巴结炎等鉴别。颈部无痛性淋巴结肿大或影像学表现为纵隔淋巴结肿大应与传染性单核细胞增多症、结核病、弓形体病、巨细胞病毒感染、白血病或NHL、胸腺瘤鉴别。局部淋巴结肿大还需要排除淋巴结炎和恶性肿瘤转移的可能。累及纵隔的HL胸部影像学表现有时与肺癌、结节病或结核相似。确诊依赖于病灶活检病理学检查。HL与NHL的鉴别要点见表16-5-2。
表16-5-2 霍奇金淋巴瘤与非霍奇金淋巴瘤的比较
续表

病理学检查发现R-S细胞对HL的诊断有重要价值,但近年研究发现R-S细胞也可见于传染性单核细胞增多症、结缔组织病及其他恶性肿瘤,因此在缺乏HL其他组织学特征,仅见到R-S细胞不能肯定诊断。Castleman病是另外一个需要与HL鉴别的疾病,(参见本章第三节“非霍奇金淋巴瘤”)。假性淋巴瘤是一种罕见的组织学上以成熟淋巴细胞组成生发中心和滤泡的慢性炎症性疾病,病理学检查是其诊断依据。
形态学和免疫组化检查有助于不同类型淋巴瘤的鉴别诊断,如富含T细胞的弥漫大B细胞淋巴瘤(DLBCL)CD30和CD15阴性,CD20和CD45阳性,可与经典型HL相鉴别,但与结节型淋巴细胞优势型HL的鉴别困难,因为两者均CD30和CD15阴性而CD45阳性,主要根据形态学特征进行鉴别。从临床和组织学特征上鉴别霍奇金淋巴瘤和原发纵隔大B细胞淋巴瘤比较困难,后者从遗传学上与经典型霍奇金淋巴瘤相似,WHO(2008)分类标准将这个“灰区”淋巴瘤暂定为一个病理实体(entity);混合细胞性霍奇金淋巴瘤由不同的细胞和基质成分组成,应与外周T细胞性淋巴瘤鉴别。总之,一般采取适当的组织病理学和免疫组化检查可以有效地鉴别,在多数情况下诊断困难是由于组织取材不足或不当所致。
【临床分期】
HL病变扩散倾向于侵犯邻近部位的淋巴结,跳跃式播散较少见,据此建立了Ann Arbor分期法。现常用Cotswold改良的Ann Arbor分期系统(表16-5-3),脾和咽淋巴环分别作为一个淋巴结区。Ⅳ期为结外病变,主要为骨髓、肺、骨骼或肝脏受累;HL累及其他部位者,需要考虑诊断是否成立或寻找有无HIV感染的证据。结合症状、体征、影像学检查和单侧骨髓活检等进行分期,根据有无全身症状分组。准确分期有助于合理治疗和预后判断。
【治疗与预后】
(一)经典型霍奇金淋巴瘤的治疗
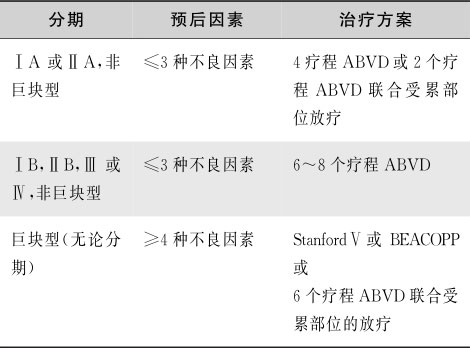
大多数HL预后较好,甚至可以治愈。选择治疗方案需考虑最大限度地减少治疗相关的远期并发症。通常根据患者的预后合理地选择治疗,35%的HL患者在确诊时病变局限,临床分期为Ⅰ期或Ⅱ期、非巨块型、无B症状、治愈率达90%~95%。Ⅲ期或Ⅳ期、巨块型、伴有B症状提示疾病处于进展期。该期患者7项独立的预后危险因素:男性、年龄大于45岁、Ⅳ期、血红蛋白<105g/L、白细胞计数>15×10 9 /L、淋巴细胞计数<0.6×10 9 /L或占白细胞的比例<8%、血清白蛋白低于40g/L,具有上述临床特征3项以下者,5年无进展生存率70%,而有4项以上特征者5年无进展生存率低于50%。
预后不同的患者在治疗上有不同的选择(表16-5-4)。
表16-5-3 霍奇金淋巴瘤分期标准(改良的Ann Arbor分期系统)

表16-5-4 成人霍奇金淋巴瘤的治疗方案
注:化疗的剂量必须根据患者的具体情况进行个体化调整
1.病变局限的霍奇金淋巴瘤的治疗
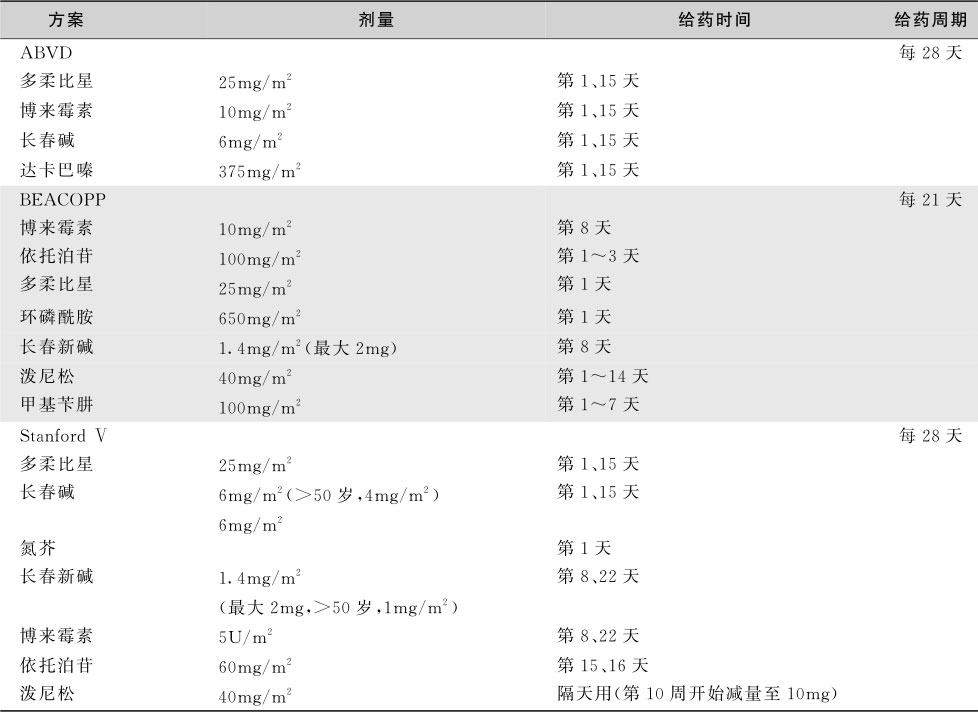
临床分期为Ⅰ期或Ⅱ期、非巨块型、无B症状。无论病变发生的部位或组织学类型,大多可以治愈。两个疗程的ABVD方案(表16-5-5)化疗联合病变部位的放疗可使95%的患者治愈,很少发生不育、过早绝经、白血病等治疗相关的并发症,主要死亡原因是放疗相关的心血管疾病及继发肿瘤。一项随机试验比较单用4~6个疗程的ABVD方案与两个疗程ABVD方案联合放疗治疗病变局限期的HL,发现单用化疗的疗效与联合放疗方案相当,在心血管疾病的发生和继发肿瘤方面两种治疗方案是否不同,还需要长期随访。该研究显示90%以上的患者只需接受4~6个疗程的ABVD方案化疗,如果经过2个疗程的化疗肿瘤不能完全缩小者,宜行PET-CT检查评估,以决定是否联合放疗,如果此时PET/CT提示仍有残留病灶者,预后相对较差。
表16-5-5 HL常用化疗方案
放疗是霍奇金淋巴瘤重要的治疗方法之一。受累野放疗是指仅仅照射病变淋巴结区域。如果患者上颈部、女性患者的腋下没有受累,应避免成为照射野。如果计划行盆腔照射,对于停经前的女性应保护卵巢功能。
2.进展期HL的治疗
也采用ABVD方案化疗作为一线治疗。联合放疗可以显著提高10年疾病无进展生存率,但不能改善总的生存,因为放疗增加了与淋巴瘤无关的其他原因所致的死亡,已取得完全缓解的患者再给予放疗无意义。化疗期间进行PET-CT检查以判断是否有淋巴瘤残留病灶,据此决定是否进行放疗对于某些患者可能有益。改良的StanfordⅤ方案和增加剂量的BEACOPP方案已被较广泛地应用于临床,这两种方案最初设计时均在化疗后对初发部位或残存的肿瘤瘤体(>5cm)进行放疗,Stanford Ⅴ方案8年无进展生存率达91%,总生存率为95%,保留了生育能力;BEACOPP联合放疗可以比COPP/ABVD方案联合放疗带来更好的无进展生存和总的生存率,但增加血液学毒性和不育症。对有0~3种不良预后因素的低危患者最常采用ABVD方案作为起始的治疗方案,70%患者有望治愈,其余30%病情仍然进展者,应给予大剂量化疗和自体造血干细胞移植。具有4种以上不良预后因素的高危患者常规化疗治愈可能性小于50%,在开始治疗时即采用StanfordⅤ或BEACOPP方案进行强烈的化疗。
3.难治或复发HL的治疗
大剂量化疗/自体造血干细胞移植(HDC/HSCT)适合以下两类患者:在初始化疗过程中或3个月内疾病继续进展(难治性HL)和完成完整的化疗疗程3个月后复发者(复发性HL)。对于仅在初发部位复发、未进行过淋巴结放疗、无B症状或结外病变者进行放疗可使40%~50%的患者治愈;对于化疗结束1年以后复发、无B症状者,再次给予化疗,联合或不联合放疗可使治愈率达到30%~40%。这两组患者在HDC/HSCT后10年无病生存率达80%。因此,对于进展期HL患者给予标准化疗疾病仍然进展者,不管复发时的特征如何,大剂量化疗后自体造血干细胞移植是标准的治疗方法。
一些新药也陆续被临床试验证实对复发、难治性HL具有一定的疗效。CD30导向抗体药物结合物brentuximab vedotin已被美国FDA批准单药治疗对于化疗和自体造血干细胞移植治疗无效的患者,具有良好的疗效和较小不良反应;此外,蛋白特异性“小分子药物”组蛋白去乙酰化酶抑制剂如帕比司他、mTOR抑制剂如依维莫司、作用于肿瘤微环境的免疫调节剂来那度胺及抗PD-1抗体等均有望成为HL治疗的新选择。
(二)淋巴细胞优势型HL(LPHL)的治疗
病程呈惰性过程,经治疗后偶有晚期复发,疾病的自然史和对治疗的反应与经典型霍奇金淋巴瘤明显不同。德国一项研究显示导致治疗失败的不良预后因素包括:分期为进展期、贫血、淋巴细胞减少,年龄(>45岁)。早期预后良好的LPHLⅠA期患者仅给予受累野照射或区域照射的5年无复发生存率95%,5年生存率100%。儿童患者在淋巴结切除后,可观察和密切随访。进展期LPHL,大多数患者可采用化疗联合或不联合放疗。常用的化疗方案有ABVD、CHOP、CVP、EPOCH方案或联合利妥昔单抗,或者单用利妥昔单抗。
【特殊情况的处理】
(一)HL伴妊娠
分期时尽量不用放射影像学检查,可采用腹部B超判断后腹膜是否存在肿块,单次胸部后前位摄片(在适当防护的前提下)用于判断纵隔是否存在巨大肿块。50%以上患者可以继续妊娠到分娩而无须治疗,如果出现症状或疾病进展,应在妊娠的中、晚期进行化疗。也可以间隙性小剂量长春碱单药化疗以控制症状,直到分娩,分娩后给予完整剂量6~8个疗程的联合化疗。
(二)HL与艾滋病
HIV感染者发生HL的风险增加了5~10倍,一般为混合细胞型或淋巴细胞削减型,易累及结外部位,尤其骨髓。80%以上患者有B症状,处于疾病的进展期,易发生机会感染,需联合抗病毒、抗真菌、粒细胞集落刺激因子等积极治疗,在此基础上给予ABVD方案或EBVP方案(表柔比星、博来霉素、长春碱和泼尼松)等化疗。但化疗药物与其他治疗药物同时使用可能降低患者的耐受性,治愈率低于非HIV感染的人群,中位生存时间一般1~2年。
(三)老年HL
预后较差,>65岁者5年生存率仅为50%,原因包括诊断时疾病已处于进展期、合并其他致死性疾病、诊断延误、分期不确切以及未维持全剂量的化疗。老年患者的治疗方案除了与年轻者相似,必要时加用中性粒细胞集落刺激因子以确保全剂量的化疗。对于存在心肺疾病者,可能需要减少或停用博来霉素或多柔比星。
【随访与远期并发症的监测】
在完成全疗程的治疗后进行全面评估,以此作为以后随访的基线,定期随访(表16-5-6)。
长期生存者,继发肿瘤、心血管疾病、甲状腺功能减退和不孕不育是最严重的迟发不良反应,随着随访时间的延长,发生率增加。
表16-5-6 霍奇金淋巴瘤化疗后的随访监测

(一)继发肿瘤
实体肿瘤是最常见的继发肿瘤,最常见于完成治疗10年后。不同一线治疗方式导致继发肿瘤的风险,从大到小依次为单纯放疗,放化疗联合,单纯化疗。单纯化疗发生肺癌或直肠癌的风险增加。肺癌和乳癌是两种霍奇金淋巴瘤治疗后最常见的继发肿瘤(占继发肿瘤的20%~25%),这可能与胸部放疗、烷化剂治疗或吸烟史等有关。对于胸部或腋下接受过放疗的女性患者每年应进行乳腺MRI或钼靶检查,至少持续到疗程结束后的8~10年,或者40岁以后。建议患者每年应到乳腺专科进行乳腺检查。
(二)心血管疾病
纵隔放疗和含有蒽环类药物的化疗是发生心血管疾病的高危因素,其中66%的致死性心脏疾患是冠心病。放疗所致的心脏毒性一般在治疗完成的5~10年后出现。心血管症状可出现于任何年龄。因此,即使没有临床症状,也建议在治疗完成10年后应随访心电图,每年监测血压。
(三)甲状腺功能减退
约50%长期生存的患者中出现甲状腺功能减退,尤其颈部或纵隔接受过放疗的患者。应对患者进行仔细的甲状腺体检,每年至少进行一次甲状腺功能检查,尤其颈部接受放疗者。
(四)肺脏毒性
已经证实含有博来霉素的方案化疗可发生博来霉素诱导的肺毒性(BPT),发生的危险因素包括年龄较大、博来霉素的累积剂量、肺部照射及既往肺病史。也有文献报道应用造血细胞生长因子增加肺毒性的发病率。
(五)生育影响
年轻HL患者的生育受影响是严重影响生活质量的治疗相关长期并发症之一。接受含有烷化剂如环磷酰胺或甲基苄肼治疗或者接受盆腔或睾丸放疗者,原发性卵巢功能不全、少精子/无精子症的发生率较高。疾病处于进展期和年龄大于30岁是女性不孕症的危险因素。推荐男性患者在治疗前冻存精子。目前尚无评估和保护女性生育功能的标准方法,在化疗前冻存卵子是保护生育功能的可选方法,但目前仍然是一种试验性手段。
主要参考文献
1.Goldman L,Schafer AI.Goldman-Cecil Medincine.25th ed.Philadelphia:Elsevier Saunders,2016,1268-1273.
2.Graf SA,Gopal AK.Treatment of relapsed classical Hodgkin lymphoma in the brentuximab vedotin era.Hematology Am Soc Hematol Educ Program,2014,(1):151-157.
3.Johnson P,Mc Kenzie H.How I treat advanced classical Hodgkin lymphoma.Blood,2015,125(11):1717-1723.
4.Stephanie S,Andreas E,Marcus R,et al.Lymphoma/Pa-thology,Diagnosis,and Treatment.2nd ed.Cambridge,United Kingdom:University Printing House,2014,61-86.
第三节
非霍奇金淋巴瘤
陈波斌
【流行病学】
非霍奇金淋巴瘤(non Hodgkin Lymphoma,NHL)占所有新诊断癌症的4%,在癌症死亡的构成也占4%。在美国经年龄调整的NHL的年发病率男性为23.2/10万,女性为16.3/10万,随着年龄的增加,发病率显著增加,诊断时的中位年龄为60岁,Burkitt淋巴瘤和淋巴母细胞淋巴瘤好发于年轻人。NHL的发病率在美国、欧洲和澳大利亚最高,而在亚洲的发病率也呈升高趋势,我国淋巴瘤的病理类型分布与欧美国家有所不同,NHL约占90%,其中T细胞型占20%~25%,B细胞型占80%左右。
【病因学】
目前病因不明。NHL的危险因素包括:
1.免疫缺陷
分为先天性和获得性,前者包括严重联合免疫缺陷症、X连锁淋巴增殖性疾病(存在 SH2D1A 基因突变,该基因编码的蛋白调节宿主应对受EBV病毒感染的细胞的免疫反应,可发生致命性传染性单核细胞增多症或霍奇金淋巴瘤)、Klinefelter综合征、Chediak-Higashi综合征、Ataxia telangiectasia综合征、Wiscott-Aldrich 综合征等;获得性免疫缺陷病如实体器官移植发生淋巴增殖性疾病者在20%以上,类风湿关节炎发生NHL的风险增加2倍,Sjögren综合征发生边缘区淋巴瘤的风险增加了30~40倍,桥本甲状腺炎、自身免疫性疾病应用甲氨蝶呤治疗者甲状腺淋巴瘤的风险增加。
2.感染
HIV感染者发生NHL的风险增加100倍以上。此外几乎所有艾滋病伴发的中枢神经系统淋巴瘤和约50%其他类型淋巴瘤与EB病毒感染有关;EB病毒与大多数移植后淋巴增殖性疾病也有一定关系。在95%以上的地方性Burkitt淋巴瘤和约40%散发性Burkitt淋巴瘤患者的细胞内可检出EB病毒基因组。在所有成人T细胞白血病/淋巴瘤患者均可检出人T淋巴细胞病毒Ⅰ型(HTLV-Ⅰ),感染 HTLV-Ⅰ者约3%有发生淋巴瘤的风险,而在感染流行区,50%以上NHL患者发病可能与HTLV-Ⅰ有关。人类疱疹病毒8(HHV-8,卡波西肉瘤相关疱疹病毒)与免疫缺陷者发生原发渗出性淋巴瘤及多中心型Castleman病有关,原发渗出性淋巴瘤患者常常同时感染了EB病毒。流行病学证据显示丙型肝炎病毒与淋巴浆细胞淋巴瘤和脾边缘区淋巴瘤有关,病毒抗原长期刺激可能导致恶性B细胞克隆出现。幽门螺杆菌与胃结外边缘区黏膜相关淋巴组织(MALT)淋巴瘤发病有关。已经证实博氏疏螺旋体与皮肤边缘区B细胞淋巴瘤有关。也有证据显示鹦鹉热衣原体与眼睛附件淋巴瘤、空肠弯曲菌与免疫增生性小肠病有关。
3.职业与环境因素
杀虫剂、有机溶剂、染发剂、紫外线、吸烟等与NHL的发病有一定的关系。农药尤其苯氧基除草剂如2,4-苯氧代乙酸与NHL有很强的联系。高脂饮食可能与NHL发病有关。霍奇金淋巴瘤经过治疗后发生NHL的风险增加大约20倍,大量吸烟者发生滤泡型淋巴瘤的风险增加。乳糜泻与肠病性T细胞淋巴瘤有关。
4.遗传因素
NHL患者的同胞、淋巴瘤或其他血液肿瘤患者的第一代亲属患NHL的风险轻度增高。肿瘤坏死因子(308G→A)、白细胞介素-10(3575T→A)及其他多态性与弥漫大B细胞淋巴瘤有关。
【病理学特征】
NHL源于免疫系统的细胞在分化的不同阶段发生突变而致。NHL的淋巴结病变常表现为多中心起源或越过邻近淋巴结向远处淋巴结跳跃式播散。侵袭性NHL常原发累及结外淋巴组织,发展迅速。NHL的淋巴结其切面外观呈鱼肉样。镜下正常淋巴结构破坏,淋巴滤泡和淋巴窦可以消失。增生或浸润的淋巴瘤细胞成分单一、排列紧密。不同来源的NHL细胞的免疫表型、染色体核型、受累基因及临床表现不同,从而形成各种NHL亚型。正常免疫细胞转化为恶性淋巴瘤细胞可能存在特定的遗传学和分子遗传学异常,部分NHL亚型原癌基因的激活与特定的遗传学异常有关。2008年WHO淋巴肿瘤分类结合了形态学、免疫学、遗传学和临床的特征,将NHL分为B细胞肿瘤、T/NK细胞肿瘤两大类(表16-5-7)。
表16-5-7 世界卫生组织(WHO)关于淋系肿瘤的分类(2008)

续表

B细胞肿瘤的各亚型由未受抗原作用的B细胞(B-naïve细胞)受到抗原作用后增殖分化直至浆细胞的各个分化阶段B细胞恶变而成,往往伴免疫缺陷或自身免疫性疾病。B细胞抗原如 CD10、CD19、CD20、CD22、CD79等,在各种B淋巴细胞肿瘤中均有不同程度的表达。在欧美国家B细胞来源的淋巴瘤占85%~90%,最常见的类型是弥漫大B细胞型淋巴瘤,占所有NHL 31%;滤泡型淋巴瘤占22%,而我国弥漫大B细胞型淋巴瘤占37.5%,滤泡淋巴瘤占9%。
T和NK细胞肿瘤在亚洲多见,其免疫表型虽可以提示恶性程度,但不是分型的依据。TCR基因重排可评价细胞的克隆性,但与亚型无关。T细胞和NK细胞肿瘤亚型的确定主要依据临床特点和病理组织细胞形态学。可分成4组:①白血病型或播散型:T-淋巴母细胞淋巴瘤/白血病、成人T细胞淋巴瘤/白血病;②结外型:NK/T细胞淋巴瘤,鼻型、肠病型T细胞淋巴瘤,肝脾T细胞淋巴瘤,原始NK细胞淋巴瘤;③皮肤型:皮下脂膜炎样T细胞淋巴瘤、蕈样肉芽肿/Sézary综合征、皮肤型间变大细胞性淋巴瘤;④结内型:外周T细胞淋巴瘤、血管免疫母细胞性淋巴瘤、全身性间变大细胞性淋巴瘤。
【临床表现】
最常见的临床表现是浅表淋巴结肿大,受累淋巴结质地韧、无触痛。纵隔或后腹膜淋巴结肿大可出现压迫或浸润症状如胸痛、咳嗽、上腔静脉综合征、腹痛、背部疼痛、脊髓压迫;输尿管受压可致肾功能不全。部分NHL可伴全身症状如发热、盗汗和原因不明的体重减轻。还可有非特征性的症状,如乏力、皮肤瘙痒等。NHL几乎可以累及任何器官而出现相应症状。骨髓受累及可全血细胞减少,表现为感染、出血和贫血。NHL也可并发各种免疫异常,如自身免疫性溶血性贫血和免疫性血小板减少性紫癜是小淋巴细胞淋巴瘤/慢性淋巴细胞白血病、弥漫大B细胞淋巴瘤以及其他亚型较常见的并发症。外周神经病变与单克隆免疫球蛋白增高有关,主要见于淋巴浆细胞性淋巴瘤。NHL相关的肿瘤伴发性综合征可影响神经系统(如脱髓鞘性多神经病、吉兰-巴雷综合征、外周神经病变等)、皮肤(如天疱疮)、肾脏(如肾小球肾炎)和多脏器损害(如血管炎、皮肌炎和胆汁淤积性黄疸)。
【诊断】
NHL的诊断依赖于肿大淋巴结或受累的器官组织活检标本的病理学检查。病变部位较深时,粗针穿刺如果取材适当,对于诊断具有一定价值;细针穿刺不应该用于淋巴瘤的诊断,它不能确定NHL的亚型。各阶段B淋巴细胞免疫表型的特点在亚型诊断中有很大的价值,免疫组化染色是分型诊断的重要依据(表16-5-8)。T细胞和NK细胞肿瘤亚型的确定主要依据临床表现和病理组织细胞形态学。细胞遗传学和分子遗传学对于疑难病例的诊断很有帮助。如t(8;14)的存在支持Burkitt淋巴瘤的诊断,而t(11;14)伴Cyclin D1的过度表达可确定套细胞淋巴瘤的诊断(表16-5-9)。一部分患者骨髓涂片中可找到淋巴瘤细胞,晚期可并发淋巴瘤细胞白血病或伴发噬血细胞综合征。总之,任何新诊断的NHL均应系统而全面的评估(表16-5-10)。
表16-5-8 常见NHL典型免疫表型
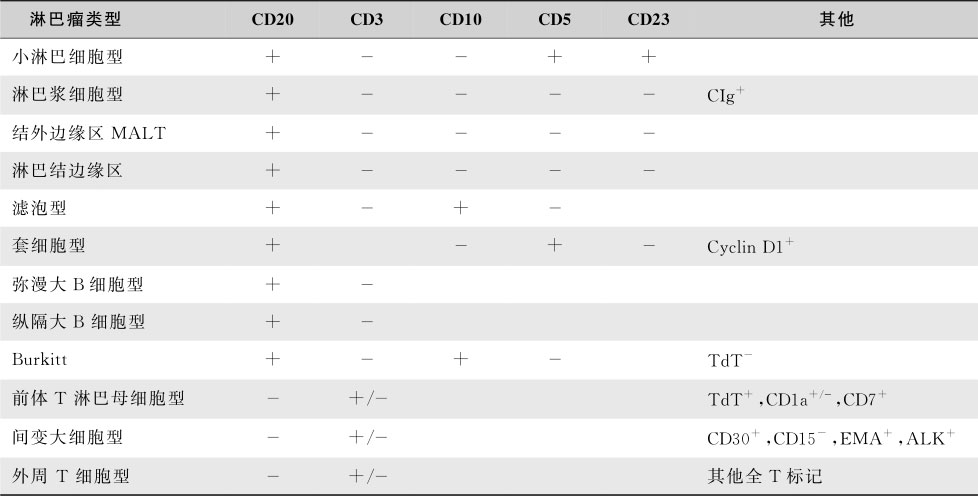
注:ALK,间变性淋巴瘤激酶;EMA,上皮细胞膜抗原
表16-5-9 NHL染色体易位特征
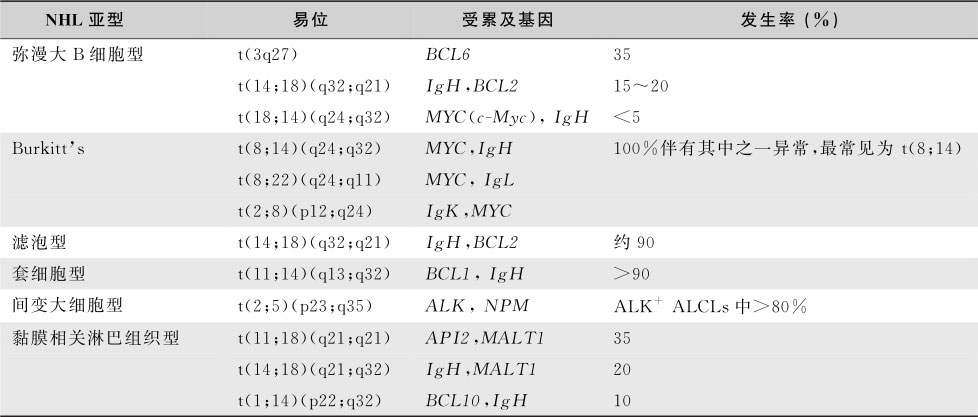
注:ALCL,间变大细胞淋巴瘤;ALK,间变细胞淋巴瘤激酶;MALT,黏膜相关淋巴组织
表16-5-10 初次诊断的非霍奇金淋巴瘤的常规评估检查

续表

【与淋巴瘤易于混淆的疾病】
(一)免疫刺激反应所致的不典型淋巴细胞增生
最常见的是多种自体免疫性疾病(如Sjögren综合征、系统性红斑狼疮、类风湿关节炎)和感染(如巨细胞病毒、EB病毒、猫爪病等)可出现B细胞和T细胞弥漫增殖伴滤泡增生,引起淋巴结肿大和淋巴结病变,易与淋巴瘤混淆,但通过病理活检及免疫学和分子生物学检查一般可以明确;仍然不能确诊者,应该对患者密切随访观察,必要时多次活检。
IgG4相关性疾病(IgG4-related disease,IgG4-RD),又称IgG4 阳性多器官淋巴增殖综合征(IgG4-positive multiorgan lymphoproliferative syndrome,IgG4 + MOLPS),是近年来新发现的疾病体,以血清中IgC4水平升高、受累组织IgG4阳性浆细胞浸润及受累组织纤维化、阻塞性静脉炎为特征。该病可累及全身多个组织器官,其中胰腺、泪腺和涎腺的受累最为常见。受累器官因纤维化、慢性炎症等出现增生肿大。患者经常伴有血清球蛋白、IgG水平升高,部分患者免疫学指标如抗核抗体及类风湿因子也可出现异常。IgG4相关性疾病的诊断标准:受累组织和器官出现弥漫性或特征性肿大、肿瘤、结节、肥厚的表现或伴功能障碍,并需满足以下指标:①血清IgG4水平升高>1.35g/L;②受累组织或器官IgG4阳性浆细胞浸润明显(IgG4 + /IgG + 细胞>40%)。
(二)非淋巴瘤的淋巴增殖性疾病
1.Castleman病(Castleman disease)
是一种原因不明的反应性淋巴结病,表现为局部巨大淋巴结,常伴发热、盗汗、体重减轻和乏力等。伴有全身症状的Castleman病往往与IL-6的过度产生有关,HIV感染者出现Castleman病常与HHV-8感染有关。临床表现有局灶型和多中心型,确诊依据病理,有透明血管型和浆细胞型。部分病例可转化为淋巴瘤。透明血管型和局灶型无全身症状,多中心型和浆细胞型常伴全身症状及多器官受累,预后不及前者。病变局限者可行手术切除或放疗。有全身病变者应用大剂量糖皮质激素可能有效。过度产生IL-6者,应用IL-6抗体常常有效。如果其他方法无效,患者接受联合化疗、自体或异基因造血干细胞移植有时有效。
2.Rosai-Dorfman综 合 征(Rosai-Dorfman syndrome)
也称为窦性组织细胞增生伴巨大淋巴结病,是一种良性局限性淋巴结增生,表现为儿童和青年出现无痛性巨大淋巴结。其病理特征为淋巴样增殖,伴较厚的纤维包膜,淋巴窦膨胀,浆细胞聚集和体积较大,常为不典型的组织细胞增殖,淋巴窦内充满吞噬性组织细胞。疾病具有自限性,可伴自身免疫性溶血性贫血,肿大淋巴结可在数周至数月内自行消退。
3.Kikuchi病(Kikuchi disease)
又称组织细胞增生性坏死性淋巴结炎(histocytic necrotizing lymphadenitis),病因不明,最常发生于年轻女性。最常见的症状包括疼痛性颈部淋巴结肿大,常伴有发热、流感样症状和皮疹。对症治疗,症状一般在数周至数月缓解。淋巴结活检显示有坏死性组织细胞灶。肾上腺皮质激素疗效甚佳。
【临床分期】
病理确诊后应进一步检查肿瘤累及的范围,以明确疾病的分期和判断预后,为选择治疗方案及疗程提供依据。除详细询问病史与仔细体格检查外,还需要进行实验室的检查,包括血常规、血清乳酸脱氢酶和β 2 -微球蛋白、骨髓涂片和活检病理学检查等;影像学检查包括胸、腹、盆腔增强CT等。正电子发射计算机体层显像CT(PET-CT)可以显示淋巴瘤病灶及部位,优于CT及其他影像学检查,可提供全身病变的完整信息,无论在肿瘤的分期还是判断疗效、预后方面同样有重要价值。高SUV提示预后较差。当SUV>13常提示为侵袭性淋巴瘤。在放射性核素浓集的部位进行穿刺可以大大提高活检病理诊断的阳性率。NHL的分期采用Cotswold改良的Ann Arbor分期系统,(参见本章第二节“霍奇金淋巴瘤”)。
【治疗】
不同组织学类型的NHL生物学特征存在差异,肿瘤的生物学行为还与病变的部位、肿块大小及患者的体能状态等有关。因其多中心发生的倾向使其治疗策略应以化疗为主。手术对于NHL的治疗作用不大。放疗常单独或与化疗联合应用于病灶局限的NHL,有时用于巨块型NHL化疗后的巩固治疗,也可用于淋巴瘤复发部位的照射以缓解症状。
(一)化疗
1.惰性淋巴瘤
B细胞惰性淋巴瘤包括小淋巴细胞淋巴瘤、浆细胞样淋巴细胞淋巴瘤、边缘区淋巴瘤和滤泡细胞淋巴瘤等;T细胞惰性淋巴瘤指蕈样肉芽肿/Sézary综合征。惰性淋巴瘤发展较慢,化、放疗有效,但不易缓解。Ⅰ期和Ⅱ期患者放疗或化疗后存活可达10年左右,部分患者可自发性肿瘤消退。Ⅲ期和Ⅳ期患者化疗后虽可能多次复发,但中位生存时间也可达10年。故在疾病早期主张观察和等待的姑息性治疗原则。若病情出现进展,可单药苯丁酸氮芥治疗,联合化疗可用CVP方案或CHOP方案(表16-5-10)。疾病进展不能控制者可试用FC方案。
2.侵袭性淋巴瘤
B细胞侵袭性淋巴瘤包括套细胞淋巴瘤、弥漫大B细胞淋巴瘤和Burkitt淋巴瘤等,T细胞淋巴瘤除了皮肤型这一组外大部分均为侵袭性。侵袭性淋巴瘤不论分期均应以化疗为主,对化疗残留肿块、局部巨大肿块或中枢神经系统累及可行局部放疗扩野照射作为化疗的补充。
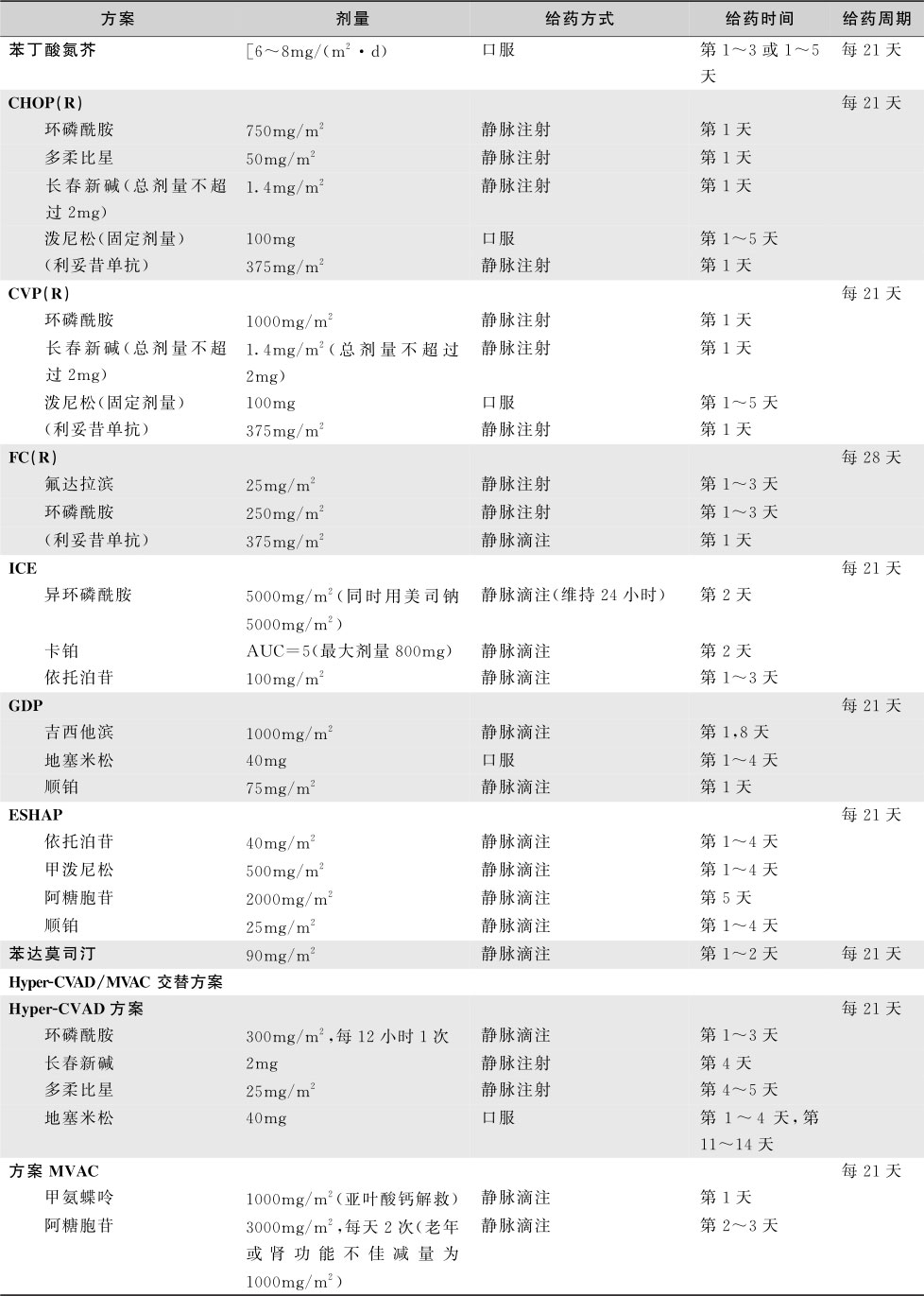
CHOP方案(表16-5-11)与其他化疗方案比较,疗效高而毒性较低,为侵袭性NHL的标准治疗方案。5年无病生存率达41%~80%。挽救性治疗可选用ICE方案、GDP方案或ESHAP方案等,对淋巴母细胞淋巴瘤/白血病、Burkitt淋巴瘤等高度恶性淋巴瘤,可试用治疗急性淋巴细胞白血病的化疗方案。
表16-5-11 NHL常用化疗方案
(二)生物治疗
CD20阳性的B细胞淋巴瘤可加CD20单抗(利妥昔单抗)治疗。可明显提高CR率和延长无病生存时间。在造血干细胞移植前用CD20单抗做体内净化,可减少移植后的复发。
(三)造血干细胞移植
详见第十一章“造血干细胞移植”。
【预后】
1993年建立的国际预后指数(IPI)评估系统被广泛用于评价NHL的预后。DLBCL的IPI与aaIPI危险分组与预后关系见表16-5-12。
表16-5-12 DLBCL国际预后指数分组与预后的关系
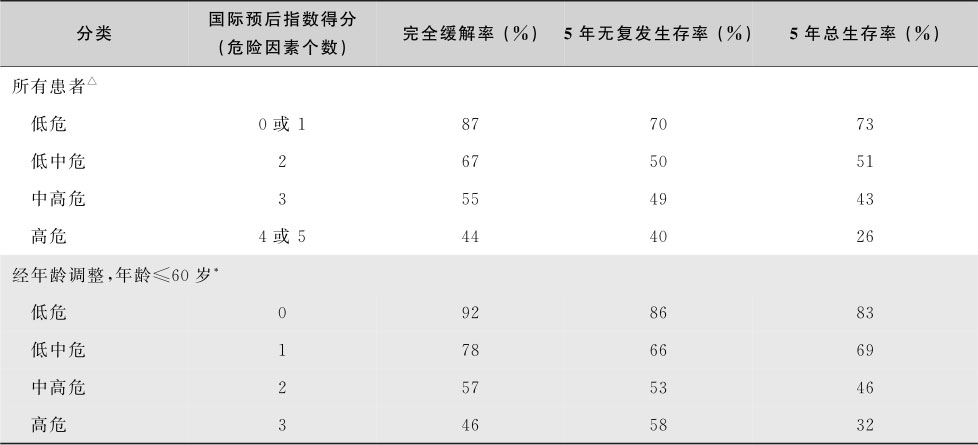
注: Δ 对于所有患者不良预后因素:年龄>60岁,LDH增高,体力状态2~4,一个以上的结外病变,Ann Arbor分期Ⅲ或Ⅳ期; * 对于≤60岁患者:LDH,增高,体能状态2~4,Ann Arbor分期Ⅲ或Ⅳ期
【部分类型淋巴肿瘤的临床特征】
(一)前体淋巴组织肿瘤
包括B淋巴母细胞性白血病/淋巴瘤和T淋巴母细胞性白血病/淋巴瘤两类。肿瘤累及淋巴结或其他实体组织,这些肿瘤细胞在形态与免疫学上与B细胞或T细胞急性淋巴细胞白血病的幼稚细胞相同。当患者有明显的淋巴结病变而骨髓病变较轻或没有累及时,被划分为淋巴母细胞性淋巴瘤,当骨髓中的肿瘤细胞比例>25%时,则诊断为淋巴细胞白血病。儿童比成人更常见。B淋巴母细胞性淋巴瘤常常表现为实体肿瘤,伴随皮肤和骨骼受累及,而T淋巴母细胞性淋巴瘤典型表现为年轻男性出现纵隔肿块。中枢神经系统受累及常见。淋巴母细胞性淋巴瘤约90%为T细胞来源,而急性淋巴细胞性白血病85%为B细胞表型。不良预后因素包括中枢神经系统受累、分期为Ⅳ期和乳酸脱氢酶升高等。
治疗可参照急性淋巴细胞性白血病的化疗方案,包括诱导、巩固、维持治疗,中枢神经系统预防。
(二)成熟B细胞肿瘤
1.慢性淋巴细胞白血病/小淋巴细胞淋巴瘤(chronic lymphocytic leukemia/small lymphocytic lymphoma,CLL/SLL)
小淋巴细胞淋巴瘤(SLL)是指淋巴结或其他组织器官被肿瘤细胞浸润,这些细胞在形态和免疫学上与慢性淋巴细胞白血病(CLL)相同,CD5 + ,CD23 + ,CD19 + ,CD10 - ,cyclin D1 - 。患者可无症状或仅有乏力等,体格检查常有淋巴结或脾大,由于存在低球蛋白血症,易于感染。如果疾病进展迅速,可出现全身症状。预后不良因素包括肿瘤细胞高表达CD38和ZAP-70、免疫球蛋白无重链基因重排、细胞遗传学异常如del(17p)和del(11q)。2%~3%的患者可转化为弥漫大B细胞淋巴瘤或霍奇金淋巴瘤(Richter综合征),一旦转化预后差。无不良预后特征的患者中位生存时间超过10年,可等待观察。对于进展迅速、出现症状性淋巴结肿大、血细胞减少者,可按惰性淋巴瘤治疗。本病不能治愈,平均发病年龄65岁,因此选择治疗方案宜个体化。氟达拉滨联合环磷酰胺和利妥昔单抗方案适用于相对年轻体能较好患者的治疗,对于老年患者则选择苯丁酸氮芥或氟达拉滨联合利妥昔单抗进行治疗。苯达莫司汀联合利妥昔单抗方案具有良好的疗效。此外,免疫调节剂来那度胺、B细胞受体通路的抑制剂依鲁替尼和idelalisib均为高效的口服药物,适于复发、难治的小淋巴细胞淋巴瘤的治疗。异基因造血干细胞移植虽可能治愈本病,但很少有患者适于移植。此外,患者可能出现自身免疫性血小板减少症、自身免疫性中性粒细胞减少症和自身免疫性溶血性贫血。这些自身免疫性疾病应用糖皮质激素、静脉用丙种球蛋白或切脾治疗有效。
CLL的治疗请参见本篇第三章第三节“慢性白血病”的相关内容。
2.黏膜相关淋巴样组织结外边缘区淋巴瘤(extranodalma rgina lzonel ymphomaofmucosa-assoc iat ed lymphoid ti ssue,MALTl ymphoma)
属惰性淋巴瘤,占B细胞淋巴瘤的7%~8%。最常见于胃肠道、腮腺、乳腺、甲状腺、眼眶、结膜、皮肤和肺。原发胃的MALT淋巴瘤约占胃淋巴瘤的50%。MALT淋巴瘤的组织学特征为异型性小B淋巴细胞,包括边缘区细胞、单核样细胞、小淋巴细胞和散在的免疫母细胞,肿瘤细胞包绕反应性滤泡,可呈星空状,肿瘤细胞侵犯上皮组织,形成淋巴上皮样病变。免疫学表型:CD20 + ,CD79 + ,CD5 - ,CD10 - ,CD23 - 。细胞遗传学可出现:t(11;18),+3等。大多数患者在确诊时为Ⅰ期或Ⅱ期。无症状患者可密切监测,有症状时治疗,局部手术或放疗可以取得很高的治愈率。30%的患者病变播散至骨髓或其他部位,呈弥漫性病变,参照滤泡型淋巴瘤治疗。胃 MALT淋巴瘤常常与幽门螺杆菌感染有关,浅表病变经抗生素治疗可使50%以上的患者获得完全缓解,但容易复发,应每3个月复查胃镜,共1.5年。如果病变侵袭较深、淋巴结播散或染色体检查提示t(11;18)存在,应予利妥昔单抗单药或联合化疗。
3.滤泡型淋巴瘤(follicular lymphoma,FL)
是一种惰性淋巴瘤,最常见无痛性淋巴结肿大,或因咳嗽、呼吸困难或胸腔积液,腹痛或腹胀发现深部淋巴结肿大就诊;少数有发热、盗汗或体重减轻等全身症状。组织学特征是有滤泡存在,外套区消失,细胞形态呈小至中等大小的生发中心细胞和大的无核裂中心母细胞组成。2008年WHO造血系统肿瘤分类标准中依据中心母细胞的数量分为1级、2级、3A级和3B级,1,2级FL呈惰性病程,不易治愈,而3级滤泡的FL有较强的侵袭性。免疫学表现:CD10 + 、CD19 + 、CD20 + 、CD79a + 、CD5 - 、CD43 - 、bcl-2 + 。细胞遗传学:几乎所有病例均有细胞遗传学异常,最常见为t(14;18)及 BCL-2 重排。3级FL参照弥漫大B细胞淋巴瘤治疗。1级和2级FL可参照惰性淋巴瘤治疗,大约5%~15%的患者在确诊时为Ⅰ~Ⅱ期,可予受累部位的放疗,10年无病生存率大约在50%以上,总的生存率在60%~70%。Ⅲ~Ⅳ期的中位生存时间为8~10年。有30%~50%可转化为弥漫大B细胞淋巴瘤。无症状者,尤其老年人和伴有其他疾病的患者,可密切随访观察。如出现全身症状或进行性淋巴结肿大、脾大、胸、腹水或血细胞减少则需要治疗。可选用利妥昔单抗375mg/m 2 静注治疗,每周1次,连续4周,可取得50%以上的有效率,疗效的中位维持时间1~2年,如果每6个月再重复以上的疗程,疗效维持的时间更长。氟达拉滨单用或联合米托蒽醌也可有效治疗FL;利妥昔单抗联合CVP方案或CHOP化疗,有效率、疗效持续时间及生存时间等均优于单纯化疗。约50%~60%复发的FL对于利妥昔单抗治疗有效,但完全缓解率只有10%。利妥昔单抗无效,应用放射性标记的单抗如托西莫单抗、替伊莫单抗(ibritumomab tiuxetan)或干扰素治疗有效。对于有症状、病变局限的患者,放疗可能有效。自体造血干细胞移植可以获得持久的缓解,而异基因造血干细胞移植可使一些复发的患者治愈。
滤泡型淋巴瘤的特殊类型包括儿童滤泡型淋巴瘤、原发小肠滤泡型淋巴瘤、滤泡内肿瘤或原位滤泡型淋巴瘤。儿童FL诊断时一般处于疾病的早期,不表达 BCL-2 ,无t(14;18),预后较成人好。原发小肠FL最近被认为是一种独立的疾病,大多发生于十二指肠,病理形态、免疫表型和细胞遗传学特征与淋巴结FL相似。多数患者的病程惰性、病变局限,即使不治疗预后也很好。原位FL定义为形态学正常的淋巴结或其他淋巴组织中有少量 BCL-2 阳性的滤泡。部分患者有FL病史或身体其他部位存在FL。也有部分患者无其他淋巴瘤的证据,这类患者不能诊断淋巴瘤,不能按照淋巴瘤治疗,推荐进行随访。
4.套细胞型淋巴瘤(mantle cell lymphoma,MCL)
套细胞型淋巴瘤是一种小淋巴细胞组成的B细胞肿瘤,占NHL的3%~10%,男性发病率是女性的2.5倍,发病率随年龄增大而增加,中位年龄68岁。多数病例均呈现进展型,60%~70%的病例在确诊时已达Ⅳ期,50%的病例有骨髓累及。结外病变以骨髓、咽淋巴环及胃肠道受累常见,尤以胃肠道多发性淋巴瘤样息肉病具有特征性,因此在确诊MCL后进行初次评估时应进行胃肠道内镜检查。组织学上肿瘤细胞为小或中等大小的淋巴细胞,形态一致,核不规则,类似有核裂滤泡中心细胞,但没有中心母细胞,淋巴结正常结构破坏。免疫学表型:sIgM + ,CD19 + ,CD5 + ,CD43 + ,CD23 - ,CD10 - ,bcl-2 + ,CyclinD1 + ,细胞 遗 传学:t(11;14)。一些患者外周血和骨髓受累及的临床表现与慢性淋巴细胞白血病相似,两者淋巴细胞均可CD5阳性,但MCL的CD23阴性。当存在t(11;14)和Cyclin D1的过度表达或FISH检查发现 BCL-1 / Ig H ( CCND1 / Ig H )易位时可以确诊MCL。套细胞淋巴瘤预后较差,中位生存时间3~4年。对于年轻或体能状态较好的患者采取较为强烈化疗(如R-Hyper-CVAD)可以改善预后,而年龄较大的患者可应用R-CHOP方案。此外,苯达莫司汀联合利妥昔单抗治疗也可获得较好的效果。一些新的药物如来那度胺、硼替佐米、依鲁替尼在临床试验中对于部分患者显示了良好的疗效。在化疗初次缓解后行自体造血干细胞移植可改善预后,但无随机临床研究证实。异基因造血干细胞移植可治愈本病,但移植相关的死亡率和病死率高。
5.弥漫大B细胞淋巴瘤(diffuse large B-cell lymphoma,DLBCL)
是最常见的NHL,约占成人NHL的30%~40%。细胞和遗传学特征存在很大的异质性。组织学特征为大B淋巴细胞弥漫浸润,细胞核为正常淋巴细胞的2倍,形态有较大变异,可分为:中心母细胞型、富含T细胞/组织细胞型、间变细胞型、免疫母细胞型;免疫学表型:sIg + 、CD19 + 、CD20 + 、CD22 + 、CD79 + 、CD5 -/+ 、CD3 - 、BCL-2 +/- 、cyclin D1 - 。近年来的基因表达谱分析可将DLBCL分为三种亚型:生发中心B细胞型、活化B细胞型、原发纵隔B细胞淋巴瘤及3型(不符合前三种亚型特征者),生发中心来源的 DLBCL(CD10 + 、BCL-6 + 、Mum-1 - )预后比非生发中心来源(CD10 - 、BCL-6 - 、Mum-1 + )的 预后好。30%以上弥漫大B细胞淋巴瘤在初诊时为Ⅰ期或Ⅱ期,目前R-CHOP方案是DLBCL的标准方案,年龄在60岁以上者,75%可取得完全缓解,5年无病生存率47%,5年总生存率58%。年轻患者采用R-DA-EPOCH方案或RACVBP可获得更好的疗效。对于具有不良预后特征的患者,在进行常规化疗达到缓解后,大剂量化疗及自体造血干细胞移植可能有益。复发难治的解救方案包含了顺铂、阿糖胞苷、依托泊苷、卡铂和异环磷酰胺,联合治疗的有效率在50%以上,但只有10%~15%的患者可取得长期无病生存。对于复发的DLBCL给予大剂量化疗和自体造血干细胞移植是公认的挽救性治疗措施;一些新药如来那度胺、硼替佐米、依鲁替尼等用于治疗复发、难治的活化B细胞性DLBCL具有较好的疗效。
6.原发纵隔大B细胞淋巴瘤(primary mediastinal large B-cell lymphoma,PMLBCL)
起源于胸腺,最常见于年轻女性,是DLBCL的一种亚型,具有独特的临床和形态学特征,与其他弥漫大B细胞淋巴瘤在遗传学上明显不同,而与经典型霍奇金淋巴瘤相似。突出的表现是纵隔肿块及由此而引起的咳嗽、胸痛或上腔静脉综合征。大多数患者的病变局限于颈部和胸部,出现巨块(>10cm)或恶性胸腔积液者提示预后不良。治疗方案参照DLBCL,也有文献报道R-EPOCH方案可取得更好的疗效。复发常常发生于淋巴结外区域,如中枢神经系统、肺、胃肠道、肝脏、卵巢和肾脏。
7.Burkitt淋巴瘤(Burkitt lymphoma,BL)
约占淋巴瘤的1%~2%,是一种高度侵袭性淋巴瘤,儿童和免疫抑制者如HIV感染者比健康成人更为常见。广泛累及淋巴结外区域。地方性BL最常见于居住于赤道非洲的儿童,下颌骨常受累及,发病与EB病毒感染有关。散发性BL最常见于儿童和年轻成人,表现为腹部肿块,有时肾脏、卵巢和乳腺可受累及,约1/3的患者病变累及骨髓。肿瘤细胞形态一致,中等大小B淋巴细胞,胞质嗜碱性,易见分裂象。免疫学:sIgM + ,CD10 + ,CD19 + ,CD20 + ,BCL-6 + ,CD5 - ,CD23 - 。细胞遗传学异常主要有t(8;14)、t(2;8)。该类淋巴瘤常有巨大肿块、增长迅速、肿瘤细胞对化疗极其敏感,在化疗过程中可出现肿瘤溶解综合征。应尽早治疗,一般采用包含利妥昔单抗在内的强烈化疗方案(R-Hyper-CVAD方案等)或R-EPOCH方案。R-CHOP方案疗效不佳。化疗开始前即予以水化、碱化。Burkitt淋巴瘤的治愈率>50%。
8.淋巴浆细胞性淋巴瘤(lymphoplasmacytic lymphoma,LPL)
是一种惰性淋巴瘤,常常累及骨髓、外周血和脾。存在单克隆IgM血症,也被称为Waldenström巨球蛋白血症(详见本篇第五章第六节“淋巴浆细胞淋巴瘤与Waldenstrom巨球蛋白血症”)。
9.脾边缘区淋巴瘤(splenic marginal zone lymphoma,SMZL)
是一种惰性淋巴瘤,常表现为脾大和淋巴细胞增多,浅表淋巴结肿大不常见。1/3~1/2的患者有单克隆球蛋白血症。可伴有自身免疫性溶血性贫血和ITP。外周血可出现绒毛状淋巴细胞,绒毛短,集中于细胞的两端,故称伴绒毛细胞脾淋巴瘤(SLVL)。切脾治疗可改善贫血和血小板减少。蒽环类药物单药化疗或以此为基础的联合化疗可能有效,也有报道应用干扰素治疗有效。利妥昔单抗治疗可取得良好疗效。对HCV阳性,有脾大,并且无抗病毒禁忌证的患者应予以抗HCV治疗。
10.淋巴结边缘区淋巴瘤(nodal marginal zone lymphoma,NMZL)
是一种惰性淋巴瘤,常出现全身性淋巴结肿大,临床过程和预后与FL相似,两者的治疗也相似。
11.血管内大B细胞淋巴瘤(introvascular large B-cell lymphoma)
是一种少见结外弥漫大B细胞淋巴瘤,肿瘤细胞浸润小血管的管腔。临床表现呈多样性,累及多脏器,可有血栓形成、出血和坏死。具有高度侵袭性,易累及骨髓,患者长期发热,极易误诊。多数患者最后由尸检确诊。
12.原发性渗出性淋巴瘤(primary effusion lymphoma)
与HHV-8病毒有关,见于HIV感染者和其他免疫缺陷患者。可出现多浆膜腔积液,无浅表淋巴结肿大,化疗疗效不佳,预后很差。
13.灰区淋巴瘤(grey zone lymphoma,GZL)
包括两型:①B细胞淋巴瘤,不能分类,具有弥漫大B细胞淋巴瘤和Burkitt淋巴瘤中间特点;②B细胞淋巴瘤,不能分类,具有弥漫大B细胞淋巴瘤和经典霍奇金淋巴瘤中间特点,灰区淋巴瘤兼有两种肿瘤的形态学和遗传学特征,但又因其临床和生物学特征的特殊性不能归入以上两种类型。建议按照预后差的类型治疗。
(三)成熟T细胞和NK细胞肿瘤
1.蕈样霉菌病/Sézary综合征(mycosis fungoides/Sézary syndrome,MF/SS)
蕈样肉芽肿(皮肤T细胞淋巴瘤)是一种惰性肿瘤,最常见于中老年人。疾病进展缓慢,不能治愈。临床分为三期:红斑期(皮损无特异性),斑块期,肿瘤期。皮肤病变的病理特点为浸润向表皮性,具有Pautrier微脓肿。肿瘤细胞为小或中等大小T淋巴细胞,细胞核呈脑回状。免疫学:CD2 + 、CD3 + 、CD5 + 、CD4 + 、CD7 - 、CD8 - 。细胞遗传学: TCR 基因重排可呈阳性。当外周血有大量脑回型核的Sézary细胞(白血病细胞),称之为Sézary综合征。疾病早期(病灶<20%体表面积)常采用紫外线照射、局部应用激素或氮芥治疗;药物干预措施包括干扰素-、类视黄醇、单克隆抗体、地尼白介素-2和细胞毒性化疗药治疗。此外,组蛋白去乙酰化药物如伏立诺他、罗米地辛,西达本胺等治疗蕈样霉菌病具有较好的疗效;自体造血干细胞移植疗效差,异基因造血干细胞移植可能对部分患者有效。
2.成人T细胞淋巴瘤/白血病(adult T-cell lymphoma/leukemia,ATLL)
与 HTLV-Ⅰ病毒感染有关,可通过哺乳、输血及血制品而传染。多数感染者无症状,但其后发生成人T细胞淋巴瘤/白血病的风险约为3%。临床可分为四型:①急性型:最常见,有全身症状,肝、脾、淋巴结肿大,皮损,白细胞升高,异常淋巴细胞>10%,高钙血症,有或无溶骨性病变;②慢性型:以皮损为主,可有肝、脾、淋巴结肿大,淋巴细胞增多,异常淋巴细胞>5%,无高钙血症,LDH增高;③冒烟型;④淋巴瘤型:有明显淋巴结肿大,外周血异常细胞<1%。急性型和淋巴瘤型肿瘤细胞有明显核多形性,分叶呈花瓣状,称花细胞。病情呈惰性进展者仅需密切观察而无需治疗。病情进展者一般需要联合化疗。5年生存率低于10%。
3.CD30阳性皮肤淋巴增殖性疾病(CD30 positive cutaneous lymphoproliferative disorders)
这类疾病具有相同的组织学表现和相互重叠的临床表现。治疗方案取决于病变进展情况。这些淋巴瘤细胞表达CD30,但不表达间变性淋巴瘤激酶(ALK)蛋白。
(1) 淋 巴 瘤 样 丘 疹 病 (lymphomatoid papulosis,LYP):
组织学呈恶性表现的克隆性疾病,表现为皮肤红斑或与皮肤颜色一致的丘疹,病变部位出现自发性溃疡和坏死,病程持续数周,虽然患者最终可能演变为淋巴瘤,但预后良好。
(2)原发性皮肤间变大细胞淋巴瘤(primary cutaneous anaplastic large cell lymphoma,PC-ALCL):
最常发生在老年男性,5年生存率>90%,常可自发性缓解。治疗措施以局部治疗(手术或放疗)为主,广泛病变时化疗。
4.间变性大细胞淋巴瘤(anaplastic large cell lymphoma,ALCL)
是一种CD30 + 的侵袭性外周T细胞NHL。2008年WHO分类将ALCL分为ALK(anaplastic lymphoma kinase,间变性淋巴瘤激酶)阳性与阴性两类。前者好发于男性儿童,后者以中老年居多,男女发病率相仿。ALCL组织病理学检查形态表现多样化,所谓“标志”细胞(hallmark cell)特征是胞质丰富,呈弱嗜碱性或双性染色,有一偏心细胞核,呈马蹄形或肾形,常含多个较小的嗜碱性核仁。ALCL细胞生长具有内聚性,并具有向淋巴结窦和滤泡旁侵犯的倾向。肿瘤细胞免疫学特征:CD30(Ki-1) + 、CD3 + ,CD15和BCL-2表达在ALK + 患者通常阳性,而在ALK - 患者常阴性。细胞分子遗传学检查: TCR 基因重排阳性;75%ALK + 患者可检测到t(2;5)(p23;q35)及相应的 NPM-ALK 融合基因。
常表现为淋巴结肿大,皮肤、骨骼和胃肠道可受累及。一线治疗可选用CHOP±依托泊苷化疗。ALK + 的年轻患者预后良好,5年生存率70%~90%;ALK - 患者疗效差,生存期短。一些新药如抗CD30的抗体药物结合物brentuximab vedotin用于治疗复发患者,具有良好疗效;ALK抑制剂克唑替尼用于治疗表达ALK的淋巴瘤有效。
5.外周T细胞淋巴瘤,非特指型(peripheral T-cell lymphoma,NOS)
是一组高度异质性的成熟T细胞肿瘤,占PTCL病例的大多数。PTCL,NOS瘤细胞CD3阳性,CD5和CD7表达常缺失,约40%表达CD52。CD57高表达常提示预后不良。部分患者可表达CD30。PTCL,NOS患者的临床表现与侵袭性B细胞淋巴瘤相似,B症状和淋巴结外病变常见。常选用类似弥漫大B细胞淋巴瘤的方案,如CHOP±依托泊苷等方案,但预后差。达到完全缓解后采用大剂量化疗联合自体造血干细胞移植可提高无病生存(DFS)率。
6.血管免疫母细胞性T细胞淋巴瘤(angioimmuno-blastic T-cell lymphoma)
表现为全身淋巴结肿大、发热、肝脾大、皮疹和多克隆球蛋白血症;常并发自身免疫性溶血性贫血和纯红再障。淋巴结结构部分消失,小至中等大小淋巴细胞弥漫性浸润副皮质区,混有嗜酸性粒细胞、浆细胞和免疫母细胞,树枝状血管明显增生,血管内皮肿胀与间质嗜酸性物质沉积。治疗方法及疗效与外周T细胞淋巴瘤非特指型相似。
7.结外NK/T细胞淋巴瘤,鼻型(extranodal NK/T-cell lymphoma,nasal type)
常发生于淋巴结外部位,尤其见于鼻腔、腭部和鼻咽部。呈破坏性肉芽肿性病变,旧称致死性中线肉芽肿。肿瘤细胞弥漫浸润,血管中心性,呈凝固性坏死。免疫学:CD2 + 、CD56 + 、sCD3 - 、cCD3 + 。本病呈侵袭性,放疗和联合化疗有效,但治疗反应差,生存期短。近年有报道含门冬酰胺酶(L-Asparaginase,L-Asp)的方案可显著提高疗效。
8.肝脾T细胞淋巴瘤(hepatosplenic T-cell lymphoma)
系结外淋巴瘤,肿瘤细胞中等大小,为毒性T细胞,T细胞受体型。特征性表现为脾、肝和骨髓窦状隙被肿瘤细胞浸润,导致肝脾大、全身症状和血细胞减少。淋巴结肿大不常见。典型表现为青年男性,常发生于曾接受异体移植者或有免疫缺陷者,化疗很少能缓解,预后差。
9.肠病相关T细胞淋巴瘤(enteropathy-associated T-cell lymphoma)
病因与麦胶肠病有关,典型表现为腹痛、腹泻,有时可出现肠穿孔。分为Ⅰ型和Ⅱ型,亚洲国家患者一般为Ⅱ型。化疗有效,但很快复发,预后较差。
10.皮下脂膜炎样T细胞淋巴瘤(subcutaneous panniculitis-like T-cell lymphoma)
是一种罕见的皮肤T细胞淋巴瘤,肿瘤细胞为多形性T细胞,在皮下组织浸润,伴反应性巨噬细胞增生,类似结节样脂膜炎,表现为多发皮下结节,可形成溃疡,常被误诊为脂膜炎。病变播散的患者可出现发热、肝脾大、全血细胞减少和噬血细胞综合征。应用治疗DLBCL的化疗方案、干扰素和放疗有时有效,但预后欠佳。
【原发性结外淋巴瘤】
(一)原发性中枢神经系统弥漫大B细胞淋巴瘤(primary DLBCL of CNS)
原发性中枢神经系统淋巴瘤(PCNSL)约占脑部肿瘤的2%~3%,病理类型大多为弥漫大B细胞淋巴瘤。常在免疫缺陷患者(如HIV感染、器官移植后)伴发,也可发生于无免疫缺陷者,肿瘤生长迅速,数周即可出现症状,主要为颅内压增高,且常累及眼球。本病需与硬脑膜淋巴瘤、血管内大B细胞淋巴瘤以及系统性淋巴瘤累及中枢神经系统鉴别。肿瘤对糖皮质激素敏感,大剂量地塞米松常可迅速缓解症状,手术切除病灶并不能延长生存。治疗的主要措施为大剂量甲氨蝶呤(MTX)化疗,剂量每次为3~8g/m 2 ,也可与大剂量阿糖胞苷或利妥昔单抗等联合。CHOP方案无效。化疗与化/放疗联合方案相比,二者的总体生存率相同,联合治疗的无进展生存率提高;全颅照射疗效可使病灶很快缩小、消失,但脑白质病的发生率增加,尤其老年患者更需要关注。
(二)原发性睾丸淋巴瘤(primary testicular lymphoma)
最常见于60岁以上男性,占全部睾丸肿瘤的5%~9%,双侧发病10%~30%,病变可同时或先后出现。治疗方案包括睾丸切除,联合化疗,中枢神经系统预防,对侧睾丸放疗。
(三)原发性胃肠道NHL(primary GI non-Hodgkin lymphoma)
是最常见的结外淋巴瘤,约占结外NHL的40%。最常见的部位是胃(50%~60%),小肠第二位(20%~30%)。85%为B细胞,50%为DLBCL。胃部淋巴瘤很少需要手术切除治疗。一般参照DLBCL方案进行化疗,有时需要辅助放疗。小肠淋巴瘤易引起出血和穿孔,尤其化疗后肿瘤组织坏死,因此一般主张手术切除后再化疗。
(四)原发性骨淋巴瘤(primary bone lymphoma)
大多为DLBCL,少数为HL。占骨原发性恶性肿瘤的7%左右。原发病变部位以下肢长骨最常见,尤其是股骨,其次为骨盆、脊柱及颌骨。治疗建议化疗联合放疗加双磷酸盐。化疗可采用CHOP样方案。放疗总剂量不宜超过50Gy,以免发生承重骨病理性骨折。
(五)原发性乳腺淋巴瘤(primary breast lymphoma)
占结外淋巴瘤的2.2%,占乳腺恶性肿瘤的0.5%。病理类型以DLBCL最为多见,其次为MALT淋巴瘤。初诊时多数患者以单侧乳腺发病,累及双侧占7.5%~30.0%。病理诊断为DLBCL的原发性乳腺淋巴瘤治疗宜采用R-CHOP化疗,大肿块或双侧病变需中枢系统预防。
【特殊情况的处理】
(一)妊娠妇女患NHL
淋巴瘤在妊娠妇女发生的肿瘤中占第四位,其中NHL的发病率低于HL,但近20年NHL的发病率增加,可能与高龄产妇增加等有关。妊娠妇女患NHL病理学类型以弥漫大B细胞淋巴瘤最常见,病变部位多为生殖器官(乳腺、卵巢、子宫等)。妊娠时诊断为NHL,其治疗涉及临床和伦理问题,需要多学科通力合作。虽然在适当防护下进行胸片检查是安全的,但往往以B超进行腹腔和盆腔检查以替代CT,在早中孕时避免X线、MRI等检查。惰性淋巴瘤(如滤泡型淋巴瘤)可以观察等待,在中晚孕阶段,如果症状明显或疾病进展予以利妥昔单抗单用或联合CHOP或CVP方案化疗,一般对于母体和胎儿影响不大;对于侵袭性(如弥漫大B细胞淋巴瘤)或高度侵袭性淋巴瘤(Burkitt淋巴瘤)患者,如果在早孕阶段确诊,终止妊娠,参照非孕NHL患者的方案治疗。如果在中、晚孕阶段诊断该病,参照非孕的NHL患者的治疗方案可能使胎儿和妊娠妇女受影响,在治疗方案中尽可能避免应用甲氨蝶呤和脂质体多柔比星。
(二)免疫缺陷者伴发NHL
HIV感染者发生NHL的风险显著增加,大多为DLBCL或BL。艾滋病相关淋巴瘤病情进展迅速,常常累及中枢神经系统和其他少见部位如胃肠道、肛门、直肠、皮肤和软组织。对于体能状态良好的患者,在应用有效的抗病毒治疗的基础上给予化疗,可以取得无HIV感染的淋巴瘤患者同样的疗效。由于中枢神经系统受累及的风险很高,因此需要预防性鞘内注射化疗药物。器官移植后患者发生NHL的风险显著增加,组织学与发生于非免疫缺陷的淋巴瘤相似。在移植手术后数周内可发生,尤其见于那些在移植后应用强烈的免疫抑制剂的患者,淋巴瘤常累及移植的器官,结外病变常见,减量或停用免疫抑制剂可使移植后淋巴增殖性疾病减轻。由于这类淋巴瘤常与EB病毒感染有关,因此有人建议移植后使用阿昔洛韦或更昔洛韦,但疗效尚存争议。
(三)老年NHL
50%以上罹患NHL的患者年龄>60岁,一般预后不良。NHL老年患者的不良后果与患者接受化疗时的药物毒性增加、缓解率低、复发率高及死于心血管疾患或其他非淋巴瘤疾病的风险增加等有关。如果患者体能状态良好,且无其他合并症,可予常规剂量。>80岁,可考虑用半量化疗。
主要参考文献
1.Goldman L,Schafer AI.Goldman-Cecil Medincine.25th ed.Philadelphia:Elsevier Saunders,2016,1257-1267.
2.Korfel A,Thiel E,Martus P,et al.Randomized phase III study of whole-brain radiotherapy for primary CNS lymphoma.Neurology.2015,84(12):1242-1248.
3.Hunt KE,Reichard KK.Diffuse large B-cell lymphoma.Arch Pathol Lab Med,2008,132(1):118-124.
4.Choi WW,Weisenburger DD,Greiner TC,et al.A new immunostain algorithm classifies diffuse large B-cell lymphoma into molecular subtypes with high accuracy.Clin Cancer Res,2009,15(17):5494-5502.
5.Brenner B,Avivi I,Lishner M.Haematological cancers in pregnancy.Lancet,2012,379(9815):580-587.
6.Marcus R,Sweetenham JW,Williams ME.Lymphoma/pathology,Diagnosis,and Treatment.2nd ed.Cambridge,United Kingdom:University Printing House,2014,61-86.
第四节
Castleman病
陈 彤
Castleman病(Castleman disease,CD)是巨大淋巴结增生或血管滤泡淋巴结增生的一类疾病,最早于1920年被发现,1954年起被认为是一类临床病理实体。病因不明,可能跟感染和炎症有关,白介素6在CD的发病中起着重要作用。根据病变累及的范围分为局灶型和多中心型两类,病理类型可为透明血管型、浆细胞型和混合型。CD需与淋巴瘤、血管免疫母细胞淋巴结病、多发性骨髓瘤等淋巴浆细胞性疾病鉴别。
局灶型CD年轻人多见,以透明血管型为主,临床表现为巨大淋巴结,纵隔淋巴结累及多见。局灶型CD多无全身症状,手术切除后可治愈。多中心型CD患者发病年龄较大,HIV患者常因感染人类疱疹病毒8(HHV-8)而表现为多中心型。该型患者可有多系统受累表现,如发热、肝脾和淋巴结肿大、高免疫球蛋白血症、重症肌无力、肾病综合征、周围神经病变、淀粉样变、动脉炎等,可有POEMS综合征。治疗通常选用淋巴瘤的化疗方案,亦可选用CD20单克隆抗体、反应停、抗病毒治疗以及自身造血干细胞移植。
大约20%~30%的CD可进展为卡波西肉瘤或淋巴瘤。CD进展后淋巴瘤多为B细胞源性,套细胞性淋巴瘤多见。局灶型CD预后较好,多中心型CD则多为侵袭性,预后较差。
第五节
多发性骨髓瘤
刘 澎
多发性骨髓瘤(multiple myeloma,MM)是一种克隆性浆细胞异常增殖的恶性疾病。异常浆细胞及其产物导致MM患者一系列靶器官功能异常和临床表现,包括骨痛及骨折、肾功能损害、贫血、高钙血症和容易罹患感染。其他少见症状有凝血功能异常、神经损害及高黏滞血症等。
【流行病学】
MM占全部恶性肿瘤的1%;是淋巴造血系统发病率居第二位的肿瘤,具体占比略高于10%。目前美国及欧洲MM的发病率约为4~6/10万/年;男女发病率之比约为2∶1。MM是主要见于老年人的恶性肿瘤,诊断时中位年龄65岁,75%的患者诊断时超过55岁,40岁以下患者只占约2%。中国尚缺乏确切的MM流行病学数据,有学者估计中国MM发病率为每年(1.0~2.5)/10万。
【病因与发病机制】
MM的确切病因尚不清楚。有资料提示暴露于放射线可以导致MM。经历20年的潜伏期后,第二次世界大战原子弹轰炸幸存者中MM发生率较正常人群显著上升。此外,苯等有机溶剂、除草剂和杀虫剂均可能与MM发病相关。
(一)MM干细胞
随着治疗MM的新药不断应用于临床,MM患者的治疗反应及长期生存均得到明显改善。但目前治疗手段仍不能使MM得到彻底治愈,绝大多数患者最终疾病复发。这一临床经过提示MM患者体内存在具有高度增殖潜能并且对抗骨髓瘤药物不敏感的肿瘤干细胞。截至目前,关于MM干细胞的确切免疫表型尚存在争议。CD138 + 细胞可以在免疫缺陷动物体内增殖并引起骨髓瘤样溶骨性损害,显示其中可能含有MM干细胞组分;但该群细胞经连续在免疫缺陷动物体内移植数代后,失去了干细胞应有的自我复制和更新的能力。同样应用连续移植的动物模型和体外克隆形成实验,有学者证实MM干细胞表型可能为CD19 + CD27 + CD38 - CD138 - ,该群细胞可以在体内外形成 MM样肿瘤;相对于CD138 + 细胞,CD138 - 细胞对地塞米松、来那度胺和硼替佐米等MM治疗药物的敏感性明显减低。MM干细胞精确表型及增殖分化网络的进一步明晰将为根治性的MM靶向治疗提供有价值的线索。
(二)骨髓微环境
MM细胞赖以生存的骨髓微环境对疾病的发生发展至关重要。骨髓微环境中有细胞外基质和大量细胞成分,后者包括基质细胞、成骨细胞、破骨细胞、T淋巴细胞、树突细胞、内皮细胞以及其他造血细胞。MM细胞与上述细胞间的相互作用由相应受体、黏附分子和细胞因子介导。微环境的其他异常,如低氧和血管新生亢进,同样是MM发病机制的重要环节。
(三)两次打击模型(two-hit model)
几乎所有MM都由一个癌前阶段,即意义未明的单克隆免疫球蛋白血症(MGUS)演变而来;MGUS患者在其一生中进展为MM或相关浆细胞肿瘤的风险为每年1/100,这一固定速率提示了一个简单的两次打击肿瘤发生模型。从MGUS转化为MM的详细机制尚未完全揭示,已有研究显示 RAS 和 p53 基因突变、p16甲基化、 MYC 基因异常、以及其他继发性基因易位在此过程中发挥重要作用。
(四)克隆演变
高通量的基因组学技术手段已经允许研究者探索MM患者体内在初诊和疾病进展等不同时间点的肿瘤细胞克隆组分。与既往人们认定的不同,MM并不是起源于单一干细胞的均质肿瘤,而是由具有巨大遗传异质性的许多亚克隆(subclone)组成。在疾病的不同阶段,患者体内MM亚克隆的种类以及相对比例各不相同。针对自然病程特别是治疗措施驱动的MM克隆演变模式的研究,将为揭示MM发生和进展的机制、实现真正意义上的个体化治疗提供崭新的思路。
【临床表现】
(一)骨骼损害
初诊时表现为骨骼损害的MM患者比例约为80%。MM最突出的临床表现为骨痛,超过2/3的患者诊断时即以此症状为主诉。骨痛发生部位主要是背部和肋骨,四肢骨骼疼痛相对较少。跌倒或者抬举物品导致的脊柱压缩性骨折,会引起突然、剧烈的背痛。突然发生的肋骨痛并伴有局部触痛,提示肋骨骨折;尽管有时细微的骨折不能被普通X线检查发现。部分患者会因椎骨塌陷导致身高减低。
(二)贫血
约75%的MM患者在诊断时即呈现贫血。贫血发生原因多为骨髓中正常造血成分被MM细胞取代,红系造血减低;其他原因可为肾功能损害所致促红细胞生成素(EPO)水平减低。MM引起的贫血为正细胞正色素性贫血。贫血导致的乏力和虚弱可为MM患者就诊主诉。
(三)肾功能损害
初诊时,近50%的MM患者血肌酐水平升高,约20%血肌酐超过177μmol/L,部分患者需要透析治疗。MM患者的肾功能损害多表现为慢性肾功能不全,在疾病进展、脱水、应用造影剂以及误用其他肾毒性药物等情况下,也可以发生急性肾衰竭。MM患者发生肾功能损害的主要原因为轻链管型肾病和高钙血症。单克隆轻链可于远曲小管和集合小管沉积形成管型。尿中游离轻链水平与管型肾病及肾功能不全的程度直接相关。骨骼破坏导致的高钙血症也可以引起肾功能不全。肾功能损害的其他原因包括淀粉样变性、获得性Fanconi综合征和轻链沉积病。
(四)高钙血症
约15%MM患者在诊断时存在高钙血症(>2.75mmol/L)。临床表现不一,可以呈现乏力、虚弱、烦渴、多尿、便秘、厌食、恶心、呕吐、神志不清,嗜睡和昏迷。应该动态监测MM患者的血钙水平,以免导致肾衰竭等严重后果。
(五)感染
感染在MM患者中较普遍。感染发生的原因是多方面的,包括受损的抗体应答反应、正常免疫球蛋白减少、中性粒细胞缺乏以及应用含糖皮质激素的化疗方案治疗。感染可以表现为肺炎、败血症,甚至脑膜炎。肺炎链球菌和葡萄球菌曾被认为是主要感染病原体,目前国外资料显示MM患者病程中一半以上呼吸道感染由革兰氏阴性细菌引发。
(六)神经系统症状
由于椎骨旁浆细胞瘤或椎骨本身损害压迫神经所导致的神经根病(Radiculopathy)是 MM最常见的神经系统并发症,通常累及胸部及腰骶部区域。MM患者如果出现严重背痛、下肢无力或感觉异常、以及膀胱和肠道功能障碍,应警惕脊髓压迫,此症状在MM患者中出现的比例为5%~10%,须紧急处理。周围神经病变在MM中较少见,如果发生,多由淀粉样变性引起。骨髓瘤细胞偶尔浸润至脑膜。颅骨病灶有时延伸形成颅内浆细胞瘤。
(七)出凝血功能异常
出血及血栓事件在MM患者中均可发生。出血主要源于大量单克隆免疫球蛋白包被血小板影响其正常功能,血小板减少是另一个重要原因。MM患者发生深部静脉血栓甚至肺栓塞的风险增加,血栓事件有时由针对MM的治疗而引发。
(八)体征
面色苍白是MM患者体格检查最常见的发现。肝脏及脾脏肿大的比例分别为5%和1%,淋巴结肿大少见。大约5%的初诊MM患者和5%的长期随访患者可于体表触及髓外浆细胞瘤,直径由小于1cm至超过10cm不等。其他体征包括骨骼畸形、局部骨骼触痛和皮肤紫癜。
【辅助检查】
(一)外周血
初诊时,75%的MM患者呈正细胞正色素性贫血;白细胞和中性粒细胞计数多正常;约5%的患者血小板减少。血涂片中偶见幼红及幼粒细胞。由大量单克隆免疫球蛋白包被的红细胞容易发生聚集,从而在血涂片呈现典型的缗钱状改变。尽管血涂片中很少检出浆细胞,应用流式细胞术可以发现约10%的MM患者外周血中含有超过0.1×10 9 /L的浆细胞。外周血中浆细胞比例>20%或绝对计数超过2.0×10 9 /L者,称为浆细胞白血病。
(二)鉴别M蛋白
骨髓瘤细胞分泌的异常单克隆免疫球蛋白称为M蛋白(又称骨髓瘤蛋白)。血清蛋白电泳中,M蛋白可以呈现为琼脂糖凝胶上的密集条带或光密度仪中的高耸窄峰。血清免疫固定电泳可以确定M蛋白的重链和轻链类型,在鉴别M蛋白方面比血清蛋白电泳更加灵敏。部分MM患者的异常浆细胞只分泌单克隆游离轻链,又称本-周蛋白(Bence Jones proteins)。单克隆轻链通常难以由血清电泳检出,但很容易在尿蛋白电泳及免疫固定电泳中发现。尿电泳需要收集患者24小时尿液。单独应用血清蛋白电泳、血清蛋白电泳结合免疫固定电泳、联用血清和尿液的蛋白电泳及免疫固定电泳检出M蛋白的概率分别为80%、93%和97%。经由上述方法鉴定,MM中M蛋白IgG型占50%,IgA型占20%,轻链型占15%~20%,IgD型占2%,双克隆型占2%,IgM型占0.5%,IgE型罕见。M蛋白轻链κ型占65%,λ型占35%。约有3%的MM患者没有M蛋白检出,称为不分泌型骨髓瘤。
(三)血清游离轻链(Free Light Chains,FLC)
FLC检测可以定量未结合到完整免疫球蛋白中的κ或λ轻链。κ/λ比值的正常范围为0.26~1.65,该比值异常提示包含相应轻链的免疫球蛋白的克隆性增殖。相较蛋白电泳和免疫固定电泳,血清FLC检测在鉴别M蛋白方面更加敏感,越来越广泛地应用于MM的诊断、风险分层和疗效评估中。对于已经接受血清FLC检查的MM患者,尿蛋白电泳及免疫固定电泳不再是必需项目。
(四)骨髓
约96%的MM患者初诊时骨髓中克隆性浆细胞比例超过10%,中位浆细胞比例50%。判定浆细胞的克隆性可以采用流式细胞学、免疫组化或免疫荧光方法检验免疫球蛋白轻链是否存在限制性表达。譬如应用第一种方法,浆细胞的克隆性由κ/λ比值判定:κ/λ比值>4∶1提示κ轻链型浆细胞克隆性增殖;该比值小于1∶2为λ轻链型MM。骨髓活检是普通骨髓穿刺涂片的有益补充,若两种方法所获浆细胞比例存在差距,以比例较高者为准。多数 MM细胞的免疫表型为胞质Ig + ,CD38 + ,CD138 + ,CD19 + ,CD56 + ,CD45 - ;20%MM细胞 呈CD20 + ,小 部分表达CD10及HLA-DR。由于MM细胞在骨髓微环境中呈局灶性分布,约有4%的初诊患者,尽管呈现典型的活动性MM表现,但首次骨髓检查浆细胞比例不足10%。这种情况下,需更换部位再次进行骨髓检查;对可疑的溶骨性损害部位骨骼或髓外浆细胞瘤的活检同样有助于确立诊断。
(五)影像学检查
普通X线检查可以揭示溶骨性损害、骨质疏松和病理性骨折等改变。骨骼损害多见于椎骨、颅骨、胸廓、骨盆和四肢骨骼近端。放射性核素( -99 锝M)骨显像在发现溶骨性损害方面效果不及普通X线检查,不应采用。CT、MRI和PET/CT是更为灵敏地鉴别MM骨骼损害的影像学方法,在诊断和疗效判定等方面得到越来越多的应用。
(六)细胞遗传学
应用间期荧光原位杂交(FISH)可以在几乎所有MM患者中检出细胞遗传学异常。但尚未发现MM特征性的细胞遗传学改变。间期FISH目前被广泛用于MM的预后分层判定。
(七)其他
90%的MM患者正常免疫球蛋白(非受累免疫球蛋白)水平减低。血清乳酸脱氢酶(LDH)水平是MM的独立预后参数。血清β 2 微球蛋白反应肿瘤负荷,并且是对MM患者分期的关键指标。其他尚可开立的检查包括浆细胞标记指数(plasma cell labeling index,PCLI)和刚果红染色(皮下脂肪、骨髓或受累器官活检标本)等。
【诊断与鉴别诊断】
(一)诊断标准
依据美国国立综合癌症网络(NCCN,v3.2016)及国际骨髓瘤工作组(IMWG,2014)相关定义的最近更新,MM按照有无特征性临床表现和是否需要治疗分为活动性骨髓瘤(有症状骨髓瘤)和冒烟型骨髓瘤(Smoldering multiple myeloma,SMM,或无症状骨髓瘤),具体标准见表16-5-13。
表16-5-13 多发性骨髓瘤诊断标准

(二)分型
MM依照受累的免疫球蛋白类型分为IgG型、IgA型、IgD型、IgM 型、IgE型、轻链型、双克隆型以及不分泌型。上述各类型又可根据轻链类型进一步分为κ型和λ型。
(三)鉴别诊断
部分自身免疫性疾病、慢性肝病、感染和转移癌等可引起反应性浆细胞增多(reactive plasmacytosis),其浆细胞呈多克隆性且患者不能检出 M蛋白。其他需与 MM鉴别的疾病包括MGUS、华氏巨球蛋白血症、轻链淀粉样变性、孤立性浆细胞瘤、B细胞淋巴瘤以及骨转移癌等。
【分期和风险分层】
MM是高度异质性的疾病,患者生存可由数月至数年不等。通过建立完善的临床分期系统,可以精确判断患者预后,选择合适的治疗方案,并且可以实现不同临床试验结果的可信比较。因其简单有效并全部使用客观参数,2005年提出的国际分期系统(International Staging System,ISS)是目前应用最广泛的MM分期系统。ISS主要反映了MM患者体内的不同肿瘤负荷,近来有学者在ISS基础上,整合进化反映MM细胞生物学特征的指标(异常的细胞遗传学和LDH水平),建立了修正的国际分期体系(Revised International Staging System,R-ISS)(表16-5-14),以期更好地区分MM患者的预后。此外,梅奥诊所的骨髓瘤风险分层系统(Mayo stratification for myeloma and risk-adapted therapy classification,mSMART)引入更多细胞遗传学信息,有助于指导治疗方案的选择(表16-5-15)。
表16-5-14 MM分期系统
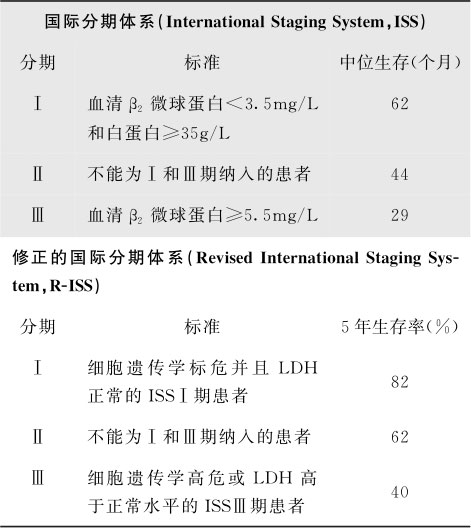
注:细胞遗传学高危指间期荧光原位杂交检出del(17p),t(4;14),t(14;16);除此之外为标危
表16-5-15 Mayo骨髓瘤风险分层系统(mSMART)
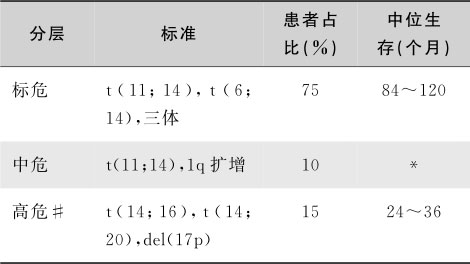
*应用硼替佐米为基础方案诱导,自体造血干细胞移植巩固并给予维持治疗,预后与标危者相仿
#在同时存在三体情况下,具有高危细胞遗传学标志的患者应被归为标危组
【疗效标准】
根据美国国立综合癌症网络(NCCN,v3.2016)及国际骨髓瘤工作组(IMWG,2014)相关定义的最近更新,可依下述标准对MM患者疗效进行判定:
完全缓解(CR):血清和尿免疫固定电泳阴性,无软组织浆细胞瘤,骨髓中浆细胞比例<5%;对仅依靠血清FLC水平作为可测量病变的患者,除满足以上CR的标准外,还要求FLC的比率恢复正常(0.26~1.65)。以上指标均需连续两次评估。
严格意义的CR(sCR):满足CR标准的基础上要求FLC比率正常以及骨髓中无克隆性浆细胞(免疫组化或2~4色的流式细胞术证实)。以上指标均需连续两次评估。
免疫表型CR(ICR):满足sCR标准的基础上,要求经多参数流式细胞术(4色以上)分析10 6 个骨髓细胞,证实无表型异常的(克隆性)浆细胞。
分子学CR(MCR):满足CR标准基础上要求等位基因特异性寡核苷酸PCR(ASO-PCR)检测阴性(敏感度要求达到10 -5 )。
非常好的PR(VGPR):蛋白电泳检测不到M蛋白,但血清和尿免疫固定电泳阳性;或血清M蛋白降低≥90%且尿M蛋白<100mg/24h;在仅依靠血清FLC水平作为可测量病变的患者,除满足以上VGPR的标准外,还要求受累和未受累FLC之间的差值缩小>90%。以上指标均需连续两次评估。
部分缓解(PR):①血清M蛋白减少≥50%,24小时尿M蛋白减少≥90%或降至<200mg/24h;②若血清和尿中M蛋白不可测,则要求受累与非受累FLC之间的差值缩小≥50%;③若血清和尿中M蛋白以及血清FLC均不可测,并且基线骨髓浆细胞比例≥30%时,则要求骨髓内浆细胞数目减少≥50%;④除上述标准外,若基线存在软组织浆细胞瘤,则要求浆细胞瘤缩小≥50%;⑤如做影像学检查,则应无新的骨质病变或原有骨质病变进展的证据。以上指标均需连续两次评估。
微小缓解(MR):血清 M蛋白减少25%~49%,24小时尿本-周蛋白减少50%~89%;若基线存在软组织浆细胞瘤,则要求浆细胞瘤缩小25%~49%;溶骨性骨骼损害的数量和大小没有增加(可允许压缩性骨折的发生)。
疾病稳定(SD):不符合CR、VGPR、PR及PD标准。如做影像学检查,则应无新的骨质病变或原有骨质病变进展的证据。
疾病进展(PD):至少应符合以下1项标准(以下百分数均为与治疗获得的相应指标最低数值相比):①血清 M蛋白升高≥25%(升高绝对值须≥5g/L),若基线血清M蛋白≥50g/L,M蛋白增加≥10g/L即可;②尿本-周蛋白升高≥25%(升高绝对值须≥200mg/24h);③若血清和尿M蛋白无法检出,则要求血清受累与非受累FLC之间的差值增加≥25%(增加绝对值须>100mg/L);④若血清和尿中 M蛋白以及血清FLC均不可测,则要求骨髓浆细胞比例升高≥25%(增加绝对值≥10%);⑤原有骨病变或软组织浆细胞瘤增大≥25%,或出现新骨骼损害或软组织浆细胞瘤;⑥出现完全归因于MM的高钙血症。在开始新治疗前必须进行连续两次疗效评估。
【治疗】
随着蛋白酶体抑制剂(硼替佐米,卡非佐米)、免疫调节剂(沙利度胺,来那度胺)等新药的应用和造血干细胞移植的广泛开展,MM患者的疗效及生存均得到明显的提高。初治活动性骨髓瘤的治疗流程见图16-5-1。
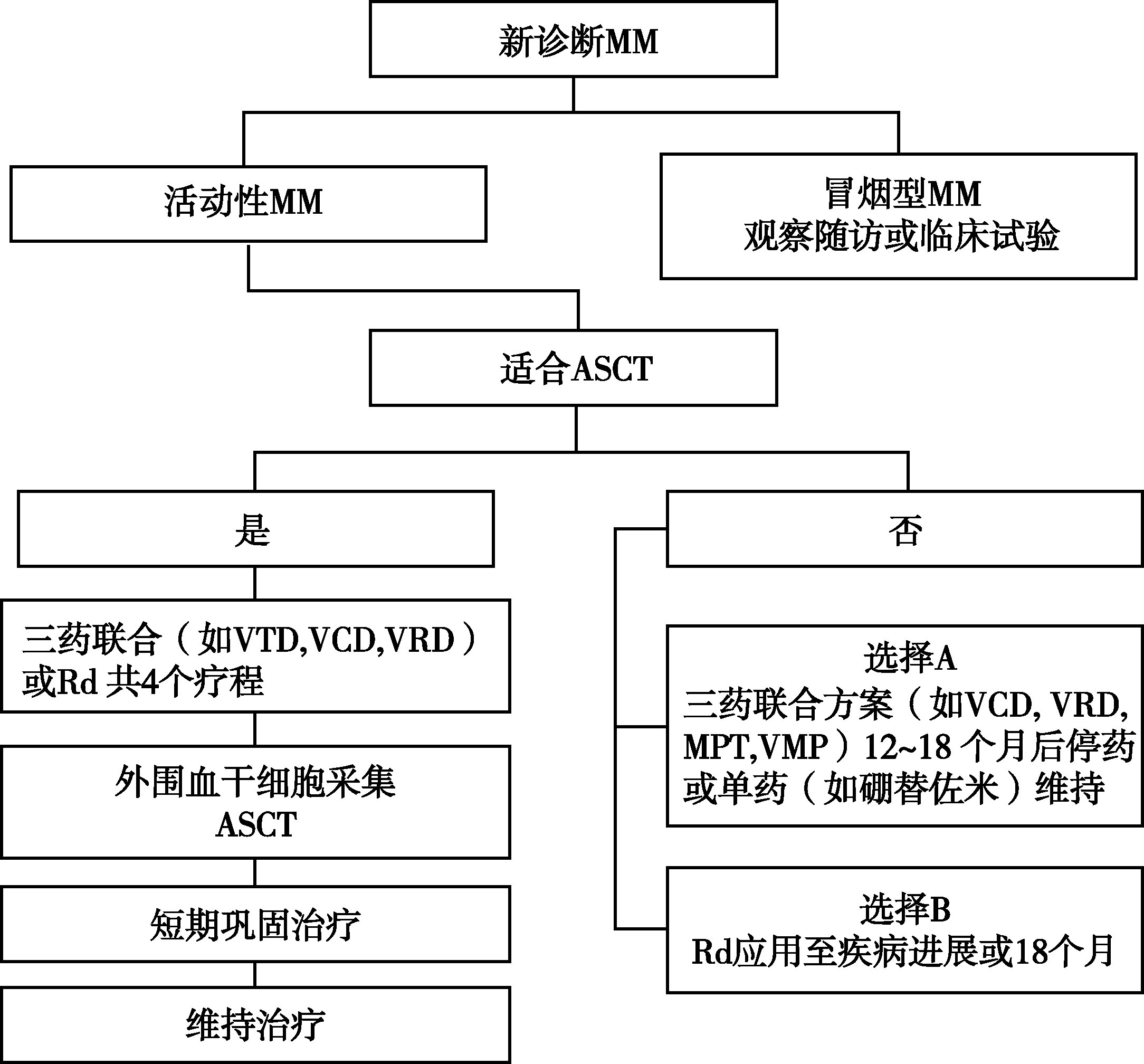
图16-5-1 多发性骨髓瘤治疗流程
(一)适合自体造血干细胞移植(ASCT)的MM患者的治疗
综合考虑体能状态评分、是否合并其他严重疾病以及年龄因素,大约有50%的初治MM患者适合接受ASCT,多数中心将移植患者年龄上限设置为65~75岁。由于目前应用的预处理方案尚不能彻底清除患者体内的MM细胞,并且回输的干细胞中有较大可能含有采集过程中混入的MM细胞,故而ASCT不能根治MM。但与单纯应用传统化疗相比,ASCT可以显著延长MM患者的无事件生存和总生存。
1.诱导治疗
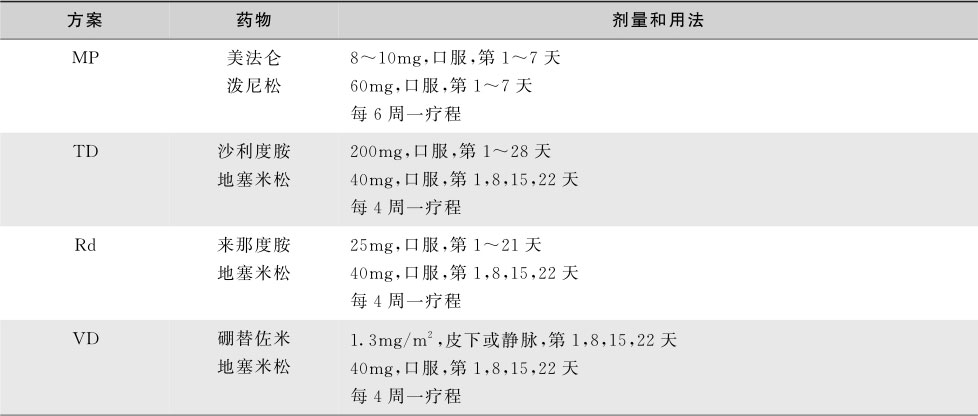
适合移植的MM的初始治疗应注意避免使用含美法仑等影响干细胞动员药物的方案。干细胞采集前通常进行4个疗程的诱导治疗。诱导方案在各国家地区因相关药物可获得性以及经济因素等有所差别。目前最常用的方案包括硼替佐米/沙利度胺/地塞米松(VTD),硼替佐米/环磷酰胺/地塞米松(VCD),硼替佐米/来那度胺/地塞米松(VRD),以及来那度胺/低剂量地塞米松(Rd)等,具体方案的剂量和用法见表16-5-16。
表16-5-16 多发性骨髓瘤常用化疗方案
续表
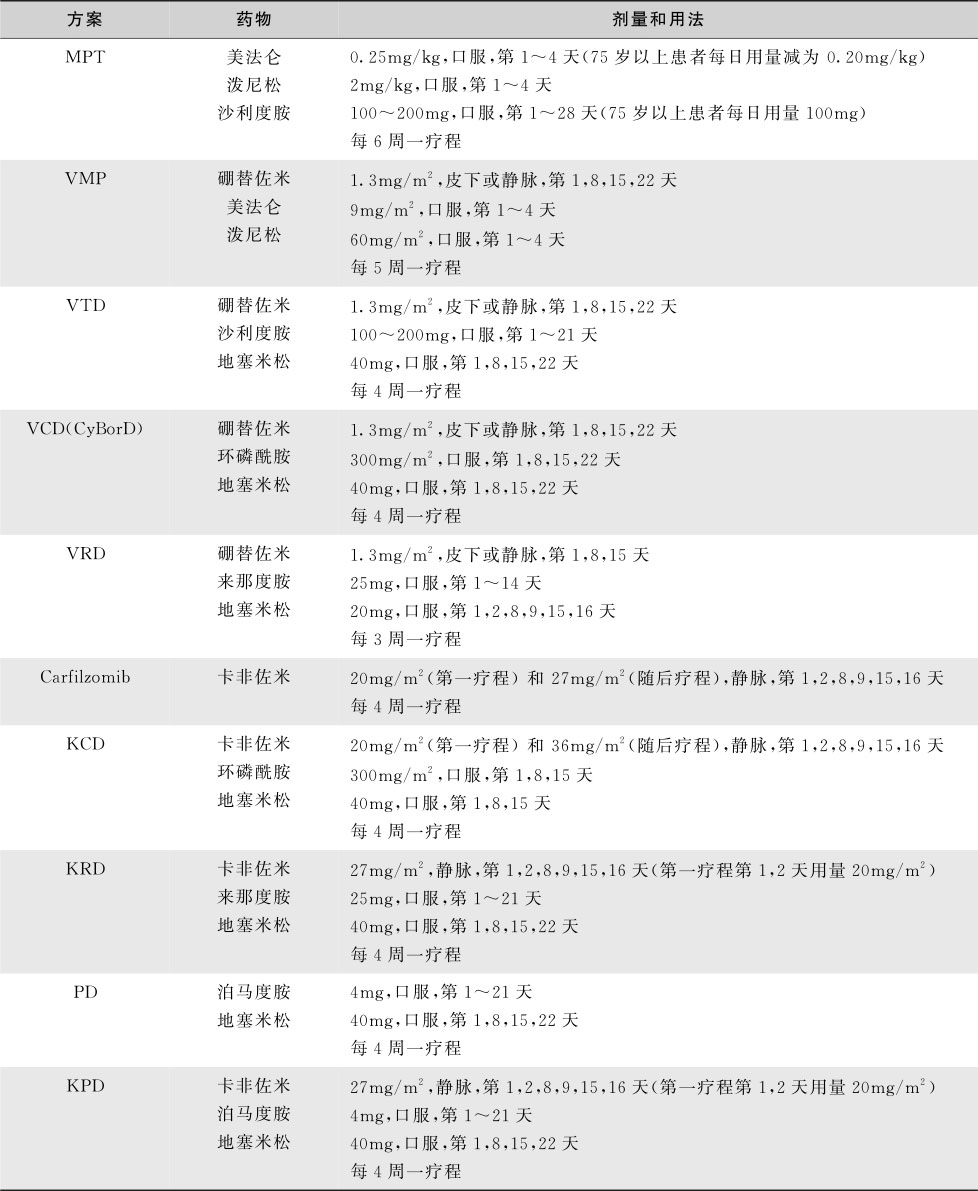
在美国东部肿瘤协作组进行的一项随机对照试验证实,在上述药物组合中低剂量的地塞米松(40mg,每周1次)相比高剂量地塞米松(40mg,第1~4天,9~12天,17~20天)给患者带来更好的总生存和更低的药物毒性。故而,在所有含地塞米松的联合方案(如VTD,VCD,VRD以及Rd等)中,均推荐应用低剂量的地塞米松(40mg,每周1次)。硼替佐米的主要副作用包括胃肠道反应、短暂的血细胞减少、乏力和周围神经病变。研究显示,相比每周两次(第1,4,8,11天)静脉给药,应用每周1次的用药间隔并采用皮下注射的给药方式能显著减低硼替佐米相关的周围神经病变。每周2次的密集给药方式,在需要紧急降低肿瘤负荷的情况下(如管型肾病引起的急性肾衰,浆细胞白血病、广泛地髓外病变和脊髓压迫等)可以考虑。沙利度胺的副作用包括镇静、乏力、皮疹、心动过缓、周围神经病变、便秘和致畸。相比沙利度胺,来那度胺的非血液学毒性显著减低,主要副作用为血小板及中性粒细胞减少。此外,有报道长期应用来那度胺会引起腹泻和腿痛性痉挛。深静脉血栓是沙利度胺和来那度胺共同的严重副作用,服用这两种药物的患者均需常规应用阿司匹林或其他抗凝剂。
2.自体造血干细胞移植
诱导治疗后应用粒细胞集落刺激因子±环磷酰胺动员采集能够满足1~2次移植的外周血造血干细胞,一份用于早期ASCT(即4次诱导方案后进行),另一份(如果采集到足够的干细胞)则冻存以备首次移植未能取得CR/VGPR或移植后复发者做第2次ASCT。常用的预处理方案是美法仑200mg/m 2 ,有学者尝试通过整合入硼替佐米或卡非佐米来改良预处理方案。与单纯常规化疗相比,ASCT能够显著提高CR率并延长MM患者的中位生存大约12个月,治疗相关死亡率1%~2%。
3.巩固与维持治疗
在ASCT后,有学者通过短期(2~4个疗程)应用与诱导阶段强度类似地联合方案(如VTD、VRD等)来提高缓解深度和对疾病的控制。另外,有报道移植后长期给予减低剂量的方案(通常单药),可以延长缓解持续时间和患者生存。一项随机对照试验显示来那度胺的维持治疗(前3个月10mg/d,如能耐受提高剂量至15mg/d)能够显著延长MM患者的无进展生存,但长期用药使得药物毒性包括致继发肿瘤的情况有增加趋势。此外,每2周1次的单药硼替佐米维持同样收到延长无进展生存和总生存的效果。考虑到长期用药毒性、经济因素、对生活质量的影响,特别在患者生存方面的获益尚待进一步证实,目前在所有患者中常规进行巩固和维持治疗尚缺乏共识。但对中危或高危的患者(推荐硼替佐米)和移植后未获得VGPR以上反应者(推荐来那度胺)可以考虑这种治疗策略。
(二)不适合自体造血干细胞移植(ASCT)的 MM患者的治疗
50%的MM患者由于各种原因无法接受ASCT。传统上对这部分患者的主要起始治疗方案为包含美法仑的组合(如 MP,MPT和VMP,具体用法见表16-5-16),三药联合方案效果优于两药组合。近来的趋势是倾向于不适合ASCT MM患者采用与适合ASCT者相同(不含美法仑)的起始方案(如VTD,VCD,VRD以及Rd等)。如采用Rd方案,建议应用至疾病进展或总共应用18个月。如采用三药联合方案(如VCD,VRD,MPT,VMP),则建议应用至12~18个月停药,或在部分中危和高危患者中尝试硼替佐米单药维持治疗。
(三)复发及难治 MM(relapsed/refractory multiple myeloma,RRMM)的治疗
尽管新药不断出现,几乎所有的MM患者终将复发,使得RRMM成为患者和临床医生面临的严重挑战。根据IMWG的定义,RRMM包括复发MM和难治MM。难治MM是指接受初次治疗或挽救治疗未获 MR以上反应甚至疾病进展(PD),以及末次治疗后60天内PD的MM患者,包括所谓“原发难治 MM”和“复发-难治MM”。复发MM为曾经接受过治疗但疾病进展需要接受挽救治疗,同时不符合前述难治MM的MM患者。RRMM治疗的策略取决于相关药物的可获得性、前期治疗的反应、复发的侵袭程度以及ASCT是否可行。
蛋白酶体抑制剂(硼替佐米,卡非佐米)、免疫调节剂(沙利度胺,来那度胺,泊马度胺)、糖皮质激素和烷化剂等单药或联合应用构成化疗方案的选择。复发表现相对惰性的MM患者可以采用Rd,VCD和VTD等方案,复发病程侵袭性较强者可采用VRD等方案。复发时呈现多发髓外浆细胞瘤或浆细胞白血病者可考虑多药联合方案,如硼替佐米+地塞米松+沙利度胺+顺铂+阿霉素+环磷酰胺+依托泊苷(VDT-PACE)。适合ASCT且前期未进行移植或ASCT后缓解时间超过18~24个月的患者(尤其是诱导治疗后已采集第二份干细胞冻存者),应考虑进行ASCT的可能。
此外,RRMM患者应积极考虑纳入临床试验。单药应用即显示出抗MM活性的新药包括MLN 9708(口服蛋白酶体抑制剂),marizomib(新型蛋白酶体抑制剂),ARRY-520(纺锤体驱动蛋白抑制剂),CD38单克隆抗体,和细胞周期蛋白依赖激酶抑制剂。可以整合入联合化疗方案的新药有panobinostat(组蛋白去乙酰化酶抑制剂)和依洛珠单抗(抗CS-1抗体)。
(四)异基因造血干细胞移植
大多数MM患者由于年龄、缺乏供者和不佳的脏器功能储备无法接受异基因造血干细胞移植。尚没有足够有力的证据显示异基因造血干细胞移植相对ASCT在疗效上的优势。考虑到现有的治疗策略的理想结果和异基因造血干细胞移植较高的治疗相关死亡率(约20%),目前认为异基因造血干细胞移植在MM治疗中的作用有限。部分年轻的高危患者在首次或第2次复发时,异基因造血干细胞移植可能是一种选择。
(五)冒烟型骨髓瘤的处理
SMM进展为活动性MM风险在诊断后的第一个5年为10%/年,第二个5年为5%/年,之后为1%~2%/年。尽管有小样本的随机对照试验显示早期治疗可以延长高危SMM患者的总生存,提早干预这部分患者的效果尚需进一步证实。目前除了临床试验,针对SMM的标准处理措施仍然是每3~4个月1次的随访观察。
【并发症处理】
(一)高钙血症
主要措施包括水化、糖皮质激素和双膦酸盐。静脉应用帕米膦酸二钠(60~90mg,维持2~4小时)或唑来膦酸(4mg,维持15分钟)可以使大多数患者血钙水平在24~72小时内恢复正常。对难治性高钙血症,可考虑应用降钙素。
(二)肾功能不全
MM患者出现急性肾衰竭主要原因为轻链管型肾病,此时多伴有血清FLC增高(血清FLC>150mg/dl),紧急处置急性肾衰关乎患者长期生存。此时最关键的措施为针对MM的有效治疗,推荐三药联合方案如VTD或VCD方案,并且药物剂量无须调整。此外,若患者尚未进入少尿期,应水化、利尿,维持尿量>100ml/h。尽管随机对照试验并未提供明确证据,仍可考虑采用血浆置换降低循环中异常轻链水平。症状性氮质血症建议血液透析,有报道提示高截留量血液透析清除FLC效果较好。高尿酸血症患者需应用别嘌醇。避免使用非甾体消炎药(NSAIDs)和静脉造影剂。长期接受双膦酸盐治疗的患者需监测肾功能。
(三)骨骼损害
防治MM骨病的关键方法是双膦酸盐的应用。每月1次静脉应用帕米膦酸二钠(60~90mg,维持4小时)或唑来膦酸(4mg,维持15分钟)可有效减低骨痛、病理性骨折和脊髓压迫的发生并延长患者的生存,全部有骨骼损害的MM患者均应接受这一预防措施。双膦酸盐的应用至少应维持1~2年,之后可减低用药频率至每3个月1次,以避免长期应用双膦酸盐引起颌骨坏死。唑来膦酸和帕米膦酸二钠均有引起颌骨坏死的报道,以唑来膦酸为多,双膦酸盐使用前应该进行口腔检查,使用中避免口腔侵袭性操作。如需进行口腔侵袭性操作,需前后停用双膦酸盐3个月,并加强抗感染治疗。
系统化疗并酌情应用镇痛剂可以使大部分的MM患者的骨痛显著缓解;仍不能减轻的剧烈疼痛且病灶局限者,可给予20~30Gy的局部放疗。椎体成形术可以缓解因严重椎骨骨折引起的疼痛。发生长骨病理性骨折以及脊柱不稳者可行外科手术治疗。对于发生病理性骨折风险较高并将严重影响生存质量和疗效的MM患者,如经审慎评估后可以考虑给予预防性的外科干预。
MM患者出现严重背痛、下肢无力或感觉异常,以及排便失禁等症状时要怀疑椎旁髓外浆细胞瘤引起的脊髓压迫。治疗措施包括地塞米松和局部放疗的应用,以及手术解压。
(四)感染
有条件的患者可以给予肺炎和流感疫苗接种。对于正常免疫球蛋白显著减低并且反复发生严重感染者,可以每3~4周静脉输注丙种球蛋白。所有接受硼替佐米或卡非佐米治疗的患者,应服用阿昔洛韦等抗病毒药物预防带状疱疹病毒激活。长期应用含糖皮质激素化疗方案的患者,应预防卡氏肺囊虫感染。
(五)其他
对接受以沙利度胺或来那度胺为基础的方案的患者,建议预防性抗凝治疗。贫血患者多数对促红细胞生成素治疗反应良好。血浆置换可显著缓解MM患者的高黏滞血症症状。
【预后】
过去20年间,随着对MM发病机制理解的深入和治疗措施的不断改进,MM患者的生存已经获得明显提升,目前中位总生存约为5年左右。在梅奥诊所,MM患者的5年总生存由1971—1996年间的37%提高到2006—2010年间的66%。但对绝大多数患者来说,目前MM仍是不可治愈的疾病。未来MM疗效和患者生存的进一步改善有赖于更多新药的研发、理想药物组合的探索以及新药与造血干细胞移植的合理整合。

(资源69) “疑难血液病临床和细胞形态学讨论”之九(病例)
主要参考文献
1.Goldman L,Schafer AI.Goldman-Cecil Medincine.25th ed.Philadelphia:Elsevier Saunders,2016,1273-1281.
2.Gertz MA,Rajkumar SV.Multiple myeloma:Diagnosis and Treatment.New York:Springer,2014,1-15.
3.RöllIgC,Knop S,Bornhäuser M.Multiple myeloma.Lancet,2015,385(9983):2197-2208.
4.Rajkumar SV,Dimopoulos MA,Palumbo A,et al.International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma.Lancet Oncol,2014,15(12):e538-548.
5.Moreau P,Attal M,Facon T.Frontline therapy of multiple myeloma.Blood,2015,125(20):3076-3084.
6.Rajkumar SV,Kumar S.Multiple Myeloma:Diagnosis and Treatment.Mayo Clin Proc,2016,91(1):101-119.
第六节
淋巴浆细胞淋巴瘤与Waldenström巨球蛋白血症
魏征 刘澎
淋巴浆细胞淋巴瘤(Lymphoplasmacytic lymphoma,LPL)是由小B淋巴细胞、浆细胞样淋巴细胞和浆细胞组成的肿瘤,通常累及骨髓,可伴有淋巴结和脾脏或其他结外器官受累,CD5通常阴性,同时不符合任何一种伴有浆细胞分化的小B淋巴细胞肿瘤诊断标准。Waldenström巨球蛋白血症(Waldenström macroglobulinemia,WM)为LPL伴骨髓侵犯及单克隆免疫球蛋白IgM血症,是LPL的主要亚型。
LPL/WM约占血液系统肿瘤1%,美国发病率约0.33/(10万人·年)。好发于老年人,男女比例1.51∶1。亚洲发病率约为美国的1/10。
【病因和发病机制】
20%~25%的LPL/WM患者与本病或其他B细胞疾病、自身免疫性疾病呈家族聚集性。
近年发现两种基因突变可能与发病有关。
1. MYD88 L256P 突变
MYD88是Toll样受体及白介素-1受体信号通路中的分子。 MYD88 L256P 突变存在于约90%的LPL/WM患者,通过激活核转录因子κB及 Bruton酪氨酸激酶(Bruton's tyrosine Kinase,BTK)信号通路促进肿瘤细胞存活。
2. CXCR4 基因突变
CXCR4 基因在30%的LPL/WM患者中发生C-末端突变,类似“疣、低丙种球蛋白血症、感染及先天性髓性粒细胞减少”综合征(Warts,Hypogammaglobinemia,Infections and Myelokathexis syndrome,WHIM综合征),故称 WHIM突变。WHIM突变通过 CXCR4 基因的配体SDF1-a调节CXCR4信号通路,促进肿瘤细胞增殖存活。
【临床表现】
老年人多见,通常累及骨髓、淋巴结及脾脏,结外受累较为少见。约1/3患者诊断时无症状。症状主要由肿瘤浸润及高IgM血症引起。
(一)血液异常
高IgM血症伴正细胞正色素性贫血常为本病首发表现。血小板减少及黏附异常、高球蛋白血症对凝血过程的干扰常可导致皮肤黏膜出血。
(二)高IgM血症
高IgM(>30g/L)可表现为视网膜病变、视力障碍、头痛、昏迷、心力衰竭等。循环IgM增多可发生冷球蛋白血症,表现为雷诺现象、关节疼痛、紫癜及皮肤溃疡等。
(三)肿瘤浸润
肿瘤主要浸润骨髓,少数患者出现髓外病变,表现为脾脏及淋巴结肿大,较少发生结外浸润。
(四)周围神经病变、冷凝集素血症
单克隆IgM可导致自身免疫紊乱,包括脱髓鞘周围神经病变及冷抗体型自身免疫性溶血性贫血。
(五)淀粉样变性
常见于单克隆IgM为λ轻链患者,轻链可沉积在心脏、肾脏、肺、淋巴结、周围神经等。
【实验室检查】
(一)电泳
大部分患者可检测到单克隆γ球蛋白血症,主要为异常增高的单克隆IgM。
(二)血液及生化
约40%患者诊断时中度贫血。重度贫血、中性粒细胞或血小板减少较少见。β 2 -MG可升高而LDH一般不升高。
(三)骨髓
骨髓涂片以小淋巴细胞、浆细胞样淋巴细胞和浆细胞浸润为主,部分患者肥大细胞比例增多。骨髓活检可见到淋巴样细胞结节样或弥漫性间质浸润,成熟浆细胞比例增高,正常造血细胞减少。
(四)免疫表型
肿瘤细胞表达全B抗原CD19、CD20、CD22、CD79a及FMC-7,单克隆膜表面Ig,很少表达CD10、CD5及CD23。
(五)遗传学异常
尚未发现LPL/WM特征性遗传学改变,FISH检测可发现超半数病例有6q-。其他常见异常包括:+18,13q-,17p异常,+4及11q异常。
【诊断与鉴别诊断】
LPL/WM主要的诊断标准包括以下两种:
(一)2003 WM国际协作组诊断标准
1.任意浓度单克隆IgM血症。
2.小B淋巴细胞、浆细胞样淋巴细胞及浆细胞浸润骨髓。
3.弥漫、间质性或结节状骨髓浸润。
4.免疫表型符合CD19+,CD20+,sIgM+;一般不表达CD5、CD10、CD23,但非排除性要求。
(二)2008 WHO LPL/WM诊断标准
1.肿瘤由小B淋巴细胞、浆细胞样淋巴细胞及浆细胞组成。
2.常累及骨髓,有时累及淋巴结和脾脏。
3.除外其他表现为浆细胞分化的小B细胞淋巴肿瘤。
4.WM:伴骨髓受累及单克隆IgM血症的LPL。
2016年新版WHO淋巴造血系统肿瘤分类中新增 MYD88 L265P 突变对诊断的作用(约90%的LPL/WM存在)。
初诊LPL/WM分期可参照其他非霍奇金淋巴瘤。需要治疗的LPL/WM患者,可通过WM国际预后评分系统(International prognosis scoring system,IPSS-WM)判断预后:①年龄>65岁;②血红蛋白≤115g/L;③血小板≤100×10 9 /L;④β 2 -MG>3mg/L;⑤血清单克隆免疫球蛋白浓度>70g/L,5个危险因素将患者分为低、中、高三组。年龄≤65岁,危险因素≤1个为低危组,2个危险因素或年龄>65岁为中危组,多于2个危险因素为高危组。
LPL/WM主要需与IgM型意义未明单克隆免疫球蛋白血症、IgM型骨髓瘤及伴有浆样分化的其他小B细胞淋巴瘤相鉴别。
【治疗】
目前LPL/WM仍是不可治愈的疾病,无症状患者可观察等待,但需严密监测评估。
(一)治疗指征及疗效评价
LPL/WM治疗指征包括临床及实验室指征。临床指征:反复发热盗汗及体重减轻;高黏滞血症;有症状的淋巴结肿大或淋巴结肿大,最大直径超过5cm;有症状的肝脾大或器官肿大、浸润;与WM相关的周围神经症状。实验室指征:有症状的冷球蛋白血症;冷凝集素血症;免疫性溶血或血小板减少;WM相关的周围神经病或淀粉样变;血红蛋白≤100g/L或血小板<100×10 9 /L。
LPL/WM疗效判断主要采用第六届国际WM工作组制定的疗效标准。
(二)血浆置换
有症状的高黏滞血症、严重的冷球蛋白血症、冷凝集素血症是血浆置换的适应证;利妥昔单抗可使IgM一过性增高,在IgM≥40g/L的无症状患者中,可在应用前行血浆置换。
(三)化疗
1.利妥昔单抗为基础
利妥昔单抗(Rituximab)无骨髓抑制及干细胞毒性尤其适合血细胞减少的LPL/WM患者,但单药疗效欠佳(30%反应率),因此需联合用药。LPL/WM患者对R-CHOP耐受较差,在血细胞减少及伴周围神经病或脏器肿大的患者一线推荐DRC方案(地塞米松、利妥昔单抗及环磷酰胺),中位PFS可达35个月;替代方案可选择苯达莫司汀联合利妥昔单抗(BR)。
2.蛋白酶体抑制剂为基础
硼替佐米可快速降低IgM,在有症状的高黏血症、冷球蛋白血症及冷凝集素血症患者中,硼替佐米为主方案有较大优势。BDR方案(硼替佐米、地塞米松及利妥昔单抗),2~3个月反应率达66~83%。二代制剂卡非佐米神经毒性较硼替佐米轻,可作为减少神经毒性的替代选择。
3.BTK抑制剂依鲁替尼(Ibrutinib,IB)
LPL中超过90%患者存在 MYD88 L265P 突变,通过BTK促进肿瘤细胞生长。BTK抑制剂有很好的抗LPL活性。在MYD88 L265P CXCR4 WT 患者中IB的总反应率为100%。
4.核苷类似物
氟达拉滨及克拉屈滨单药治疗WM有良好的反应率,无法耐受联合化疗的老年患者,可予氟达拉滨单药治疗。但因增加组织学转化及骨髓增生异常综合征风险,在年轻患者中应用受到一定限制。
5.其他
免疫调节剂沙利度胺及来那度胺分别因神经及骨髓毒性,限制了其在WM中应用;mTOR抑制剂依维莫司正在进行复发难治WM的临床试验并已取得初步疗效。
(四)移植
在化疗敏感的复发患者中,低中危患者可考虑自体干细胞移植,高危患者可考虑异基因造血干细胞移植。
主要参考文献
1.沈志祥,朱雄珍.恶性淋巴瘤.第2版.北京:人民卫生出版社,2011,493-507.
2.Gertz MA.Waldenström macroglobulinemia:2015 update on diagnosis,risk stratification,and management.Am J Hematol,2015,90(4):346-354.
3.Swerdlow SH,Campo E,Pileri SA,et al.The 2016 revision of the World Health Organization(WHO)classification of lymphoid neoplasms.Blood,2016,127(20):2375-2390.
第七节
其他浆细胞病
季丽莉 刘澎
一、意义未明的单克隆球蛋白血症
意义未明的单克隆球蛋白血症(monoclonal gammopathy of undetermined significance,MGUS)是一组单克隆球蛋白(monoclonal globulin,M蛋白)升高的良性疾病。这类患者将来发展成为多发性骨髓瘤、轻链淀粉样变、华氏巨球蛋白血症、及其他淋巴瘤的风险升高,每年有1%的患者出现进展。
MGUS在人群中的检出率是0.3%~6.6%,在不同种族分布略有不同,随年龄增加而提高,年龄调整后的发病率男性高于女性(4.0% vs .2.7%, P <0.01)。
MGUS的诊断标准是:单克隆球蛋白升高,M蛋白<30g/L;骨髓克隆性浆细胞<10%;不伴有CRAB症状(见本章第五节“多发性骨髓瘤”)。需要通过血尿免疫固定电泳、血清游离轻链(serum free light chain,sFLC)、骨髓穿刺及活检进行形态学检查和流式细胞仪检测等寻找克隆性浆细胞增殖的依据,同时排除其他疾病(恶性肿瘤、风湿性疾病、感染,以及多发性骨髓瘤、淀粉样变等)。确诊后应定期(6~12个月,以及出现相关症状时)随访,内容包括血常规、免疫固定电泳、免疫球蛋白定量、肾功能、血钙、sFLC,当出现CRAB症状时加做骨髓穿刺和骨骼影像学检查。
大部分MGUS患者没有症状,但少部分患者可出现肾脏损害,发病可能与M蛋白相关,病理类型包括膜增生性肾小球肾炎、C3肾小球肾炎和其他多种类型。MGUS对患者的主要损害在于向多发性骨髓瘤、轻链型淀粉样变、华氏巨球蛋白血症、及其他淋巴系统增殖性疾病进展的风险增大。多个指标可预测MGUS患者进展的风险:
1.Mayo标准
使用三个指标,①非IgG型 M蛋白;②M蛋白高于15g/L;③sFLC比值异常。具有0个、1个、2个、3个高危因素的患者在20年进展为MM的风险分别是5%,21%,37%,58%。
2.西班牙标准
根据两个因素:①骨髓浆细胞的免疫表型异常:流式细胞术分析,异常指CD19和(或)CD45缺失、CD38表达下调、CD56表达上调。骨髓异常浆细胞占95%以上记为1分;②骨髓浆细胞DNA非整倍体记为1分。0分、1分、2分的患者在5年进展为MM的风险分别是2%,10%,46%。
二、冒烟型多发性骨髓瘤
冒烟型多发性骨髓瘤(smoldering multiple myeloma,SMM)在临床中相对少见,在新诊断的MM中约占8%~15%。其诊断标准是:M蛋白≥30g/L,或尿 M蛋白≥500mg/24h,和(或)骨髓克隆性浆细胞10%~60%;不伴有活动性骨髓瘤症状。轻链型SMM是一种特殊的类型,仅合成克隆性FLC,而不表达免疫球蛋白重链,因此表现为尿中单克隆FLC排出过多(本周氏蛋白尿)。
SMM进展为有症状 MM的风险为:前5年,每年10%;第二个5年,每年3%~5%;10年后的进展风险是每年1%。由于其进展的风险异质性较大,需要更多的预后指标进行危险分层:
M蛋白的类型和定量:IgA型SMM较IgG型SMM进展快,中位进展时间(time to progression,TTP)分别是27个月和75个月;轻链型SMM进展风险较低,中位TTP是159个月。M蛋白定量是SMM进展的独立预后指标,血清M蛋白≥30g/L是疾病进展的独立高危因素。24小时尿M蛋白定量可预测轻链SMM的进展风险。
免疫不全(immunoparesis),指非累及免疫球蛋白低于正常,大约80%的患者会出现1或2型非累及免疫球蛋白降低,免疫不全往往与进展风险增加有关。无免疫不全、1型非累及免疫球蛋白减低、2型非累及免疫球蛋白减低的患者中位TTP分别是159个月、89个月和32个月。
sFLC比值:受累游离轻链与未受累游离轻链的比值大于8,向症状性MM进展风险显著升高,中位TTP仅30个月(sFLC比值小于8的患者中位TTP为110个月)。
骨髓浆细胞比例及免疫表型:骨髓浆细胞<20%、20%~50%、>50%(<60%)的患者进展为症状性 MM的中位TTP分别是117个月、26个月、21个月。流式细胞术分析浆细胞免疫表型,异常细胞定义为CD19和(或)CD45缺失、CD38表达下调、CD56表达上调。一项西班牙研究结果显示异常浆细胞≥95%者中位TTP仅34个月,而异常浆细胞<95%者中位TTP尚未达到。
细胞遗传学异常:具有t(4;14)、del(17p)为高危患者,进展时间最短(中位TTP 24个月),其后依次是染色体三体(中危)、其他细胞遗传学异常(标危)和无异常(低危)。基因表达谱分析(gene expression profile,GEP)也可作为SMM进展风险的评估指标,需要进一步的研究。
影像学检查:MRI是应用较为广泛的影像学检查,局限性病灶和弥散性骨髓病变是进展高危因素,全身MRI检查发现>1个局限性病灶的患者,由于2年进展为症状性MM概率达70%,目前已被归入症状性MM。PET-CT能反应病灶的活性,具有功能评判的价值。
其他指标:如M蛋白升高明显(连续两次随访中M蛋白增长≥25%)、流式细胞术检查外周血循环浆细胞>5×10 6 /L、浆细胞标记指数(plasma cell labeling index,PCLI)≥1等,都是SMM进展风险的指标。
SMM的处理仍然以随访为主,高危SMM患者可考虑参加临床试验,同时需要较密切的临床随访。随访5年未进展的SMM可降低随访频率。
三、孤立性浆细胞瘤
孤立性浆细胞瘤(solitary plasmacytoma,SP)是一种少见的浆细胞肿瘤,占所有浆细胞肿瘤的5%。可以发生在骨髓以外的任何器官,按发生部位可分为骨SP(bone SP,SBP)和髓外SP(extramedullary SP,SEP)。SBP多发于中轴骨,如脊柱和颅骨;而SEP多发生于上呼吸道,以鼻腔、副鼻窦和鼻咽部最为常见。多见于40岁以上男性,患者中位年龄是55岁,男女比例是2∶1。
诊断要求活检证实为单克隆浆细胞肿瘤,并除外MM:骨髓中克隆性浆细胞<10%(少数学者要求骨髓无浆细胞克隆性增殖),血清可有M蛋白存在,尿本周氏蛋白可阳性;需除外CRAB及其他靶器官受损症状。
SP有三种进展方式:进展为MM、局部复发和出现新病灶但达不到MM诊断标准。SBP预后较差,SBP患者10年、15年进展为MM的风险分别是65%~84%、和65%~100%。即使给予治愈性的治疗,进展为 MM的中位时间是2~3年。相比而言,SEP预后稍好,进展为 MM的患者占50%~60%,即使进展为MM,其5年生存率是100%,而SBP进展为MM的患者5年生存率仅33%。
SP治疗以局部放疗和手术切除为主要治疗。对有不良预后因素(如分化差、局部破坏、浸润明显等)患者可以考虑辅助性化疗,以减缓病情进展。化疗方案参考MM。
四、浆细胞白血病
浆细胞白血病(plasma cell leukemia,PCL)是一种侵袭性极强的少见恶性浆细胞疾病,发生率为每年每100 000人0.04例。诊断标准:外周血浆细胞增加,占白细胞的20%以上,并且外周血浆细胞绝对值>2×10 9 /L。PCL可分为原发 PCL(primary PCL,pPCL)和多发性骨髓瘤治疗后继发的PCL(secondary PCL,sPCL)。虽然两者具有外周血浆细胞升高、预后恶劣的共同特点,但是两种不同的疾病:pPCL起病急,对强烈化疗敏感,几乎100%化疗有效,但疾病常在早期复发、并迅速进展,预后不良;而sPCL是MM复发进展后的临床终末阶段,50%以上患者对治疗无效。
p PCL中位发病年龄是65岁,起病急,侵袭性强,临床上表现为严重的贫血,体检可发现肝脾大、淋巴结肿大、胸腔积液,常有中枢神经系统侵犯。溶骨性病变较MM少见(p PCL 35%,MM 80%),而髓外软组织浆细胞瘤较常见。实验室检查可以发现白细胞显著升高,乳酸脱氢酶和β 2 微球蛋白较MM患者有更明显的升高。骨髓中浆细胞浸润广泛,浆细胞呈现浆母细胞形态。pPCL肿瘤细胞表面CD27,CD56,CD71,CD117,HLA-DR 表达率较 MM 浆细胞低,而CD20,CD44,CD45,CD19,CD23表达较多。细胞遗传学和分子生物学检查往往提示PCL具有更恶劣的基因异常:del(17p),1q21扩增, MYC 异常等。
PCL一经诊断,需尽快治疗。硼替佐米可显著改善疗效,是本病治疗的主要药物。强烈化疗方案(如hyper-CVAD和DT-PACE)诱导、序贯干细胞移植可改善患者长期生存。如患者年龄小于50岁,同时具备合适供者,则建议异基因干细胞移植。对于sPCL和复发的pPCL,治疗需要考虑前序治疗方案和疾病进展的时间,多数患者可从硼替佐米和大剂量化疗中短暂获益。PCL总体预后不良,内外均缺少满意的治疗经验。
五、轻链型淀粉样变
系统性淀粉样变(systemic amyloidosis)指一组少见的淀粉样物质沉积在细胞外中形成的疾病。轻链(amyloid lightchain,AL)沉积所致系统性淀粉样变是最常见的一种,发病率约每百万人中3~5例。AL临床表现多样,缺乏特异性。约有50%患者出现心脏累及,是淀粉样变患者主要的死因。表现为限制性心肌病,通常伴有不相称的右心衰,晚期患者可表现为心脏射血分数降低和低血压。肾脏是AL型淀粉样变累及最多的脏器,表现为蛋白尿,常进展为肾病综合征,而肾功能不全仅在晚期患者出现。约1/5系统性AL型淀粉样变患者可出现周围神经轴索病变,表现为感觉异常,可伴有疼痛,很难与慢性炎症性脱髓鞘病变鉴别。自主神经也可受累,表现为直立性低血压、腹泻、便秘。软组织受累是AL型淀粉样变独特的临床表现(腕管综合征除外),常见症状有巨舌、假性肌肥大、唾液腺增大、下颌下软组织浸润。巨舌和眶周紫癜同时出现是AL淀粉样变最具特征性的表现,具有诊断意义,但仅有1/3的患者有此表现。
目前,受累器官活组织检查行刚果红染色仍是淀粉样变诊断的金标准,如受累器官活检无法操作,腹壁下脂肪活检可替代(简单易行、阳性率高)。如腹壁皮下脂肪刚果红染色阴性不能作为排除诊断的标准,可进一步行直肠或唇腺活检。新兴的诊断性辅助检查是对受累组织沉积物质的质谱分析,其意义在于明确沉积物成分以指导后续治疗,但该技术在国内尚未普及。AL型淀粉样变尚需完善免疫固定电泳、血清FLC、骨髓穿刺等寻找潜在浆细胞疾病的依据。大多AL型淀粉样变患者浆细胞疾病处于MGUS阶段,约10%~15%患者同时患有MM。
AL系统性淀粉样变患者治疗沿用多发性骨髓瘤化疗方案,如美法仑联合地塞米松、含有新药(沙利度胺、硼替佐米、或来那度胺)的化疗,低危患者可序贯自体干细胞移植。尽管新型化疗药物可显著改善AL淀粉样变的预后,但晚期患者仍然疗效不佳,早期明确诊断是改善预后的关键。
局限性AL淀粉样变与克隆性B细胞分泌淀粉样轻链局部沉积有关,常见部位有呼吸道、膀胱、眼睑和皮肤,呈惰性过程,几乎不会发展为系统性淀粉样变。局部手术治疗是主要的治疗方式,放疗可选择。
六、POEMS综合征
POEMS综合征是一种少见的副癌综合征,于2007年制定了最新的POEMS综合征诊断标准:同时具备2条必备条件(单克隆浆细胞增殖性疾病,和多发性周围神经病变),并且1条主要标准(Castleman病、VEGF升高、硬化性骨病),1条次要标准(视乳头水肿、内分泌异常皮肤改变、器官肿大、血管外液体容量增加、血小板增多/红细胞增多)。
POEMS综合征的治疗建立在对患者的全面临床评估之上:如患者仅表现为局限性骨损害、且骨髓穿刺未找到克隆性浆细胞依据,首选放疗。如患者具有2处以上骨骼病变,或找到克隆性浆细胞疾病依据,需要接受全身化疗,化疗方案有传统的美法仑联合地塞米松方案,包含新药(沙利度胺、来那度胺、硼替佐米)的化疗方案可有效提升其治疗效果。高剂量化疗联合外周血干细胞移植也是有效的治疗选择。

(资源70) “疑难血液病临床和细胞形态学讨论”之十(病例)
第八节
淋巴结肿大与脾大
陈 彤
一、淋巴结肿大
淋巴结是外周免疫器官,是淋巴细胞增殖与分化的场所。正常淋巴结很小,直径多在0.2~0.5cm之间,呈豆形或椭圆形,质地柔软,扁平,表面光滑,无压痛,与周围组织无粘连。如淋巴结直径超过1~2cm以上,外形改变,质地异常者称为淋巴结肿大(lymphadenopathy)。
健康成人浅表淋巴结除在颌下、腋窝、腹股沟可触及少数淋巴结以外,一般不易触及,如在枕后、耳后、滑车上、锁骨上等部位触及则属异常。事实上健康成人在颌下、腹股沟部位扪及1~2cm的淋巴结十分常见。深部淋巴结肿大须借影像学检查才能发现,临床上常见肺门及纵隔的气管、支气管旁及气管分叉部,以及腹膜后主动脉旁和肠系膜淋巴结肿大。儿童由于经常接触新抗原,大部分健康儿童和青少年可触及一定程度的肿大淋巴结,质地软,扁平。成人淋巴结直径>1~2cm可认为异常,而肿大淋巴结所处的区域可提示外来抗原入侵的部位。局限性淋巴结肿大(localized lymphadenopathy)是指一个引流区域的累及。全身性淋巴结肿大(disseminated lymphadenopathy)系指至少有两个以上非毗连区域的淋巴结同时肿大。
【病因】
在西方国家,大约2/3就诊于家庭医生的淋巴结肿大患者无特殊原因,肿瘤性疾病仅占1%左右。免疫活性细胞的增殖、外来的细胞和物质的浸润都可致淋巴结肿大,具体病因见表16-5-17。
表16-5-17 淋巴结肿大的病因

感染是淋巴结肿大的最常见因素,病毒、细菌、真菌、原虫等病原微生物的感染都可导致引流区淋巴结的局限性肿大,在鼠疫、猫抓病等感染性疾病中尤为突出;全身性淋巴结肿大则多见于病毒感染和弓形虫病。
淋巴结内细胞的增殖和浸润都可导致局限性或全身性淋巴结肿大,常是免疫性疾病主要临床表现,在类风湿关节炎、SLE等自身免疫性疾病常合并有淋巴组织增生和淋巴瘤的可能,增加了确诊的难度。某些良性淋巴增生性疾病,如Castleman病、药物高反应、血管滤泡性淋巴结增生症等也易与淋巴瘤混淆。而某些淋巴结肿大的疾病有明显的地域特点,如Kawasaki病、Kimura病等在亚洲多见。
淋巴结肿大的另一重要原因是免疫细胞的恶变,常是淋巴瘤、慢性淋巴细胞白血病、Waldenström巨球蛋白血症等淋巴增殖性肿瘤的首发症状,在髓系肿瘤中较为少见,多发性骨髓瘤中更罕见。在其他实体器官恶性肿瘤中,肿瘤细胞可循淋巴转移而导致相关引流区域的淋巴结肿大,晚期肿瘤也可见淋巴结广泛转移。
其他疾病,如脂质沉积病、内分泌疾病、结节病、淀粉样变性(骨髓瘤、遗传性、慢性炎性病变)等都可导致淋巴结肿大。
【诊断和鉴别诊断】
淋巴结肿大患者需重视其有无相关的全身表现和患者的年龄、既往史、肿大淋巴结的部位和范围、大小、持续时间和伴随的炎症表现(表16-5-17)。有明确的感染症状且肿大淋巴结位于病灶的淋巴引流区可考虑感染所致;有可致淋巴结肿大的自身免疫性疾病史者可考虑为原发病所致。但是,上述患者若淋巴结持续肿大仍需考虑淋巴结活检,此外,局限性的淋巴结进行性肿大,尤其伴发热、体重减轻时,需行淋巴结活检以除外淋巴瘤。
(一)病史
咽痛、咳嗽、发热、盗汗、乏力、消瘦和淋巴结疼痛以及患者的年龄、性别、职业、动物接触史、性行为和用药史等病史对淋巴结肿大的鉴别诊断很有帮助。急性淋巴结肿大、有疼痛和发热,多为感染性疾病所致,病史较长的慢性淋巴结肿大可考虑慢性感染、结缔组织病等因素。
(二)体格检查
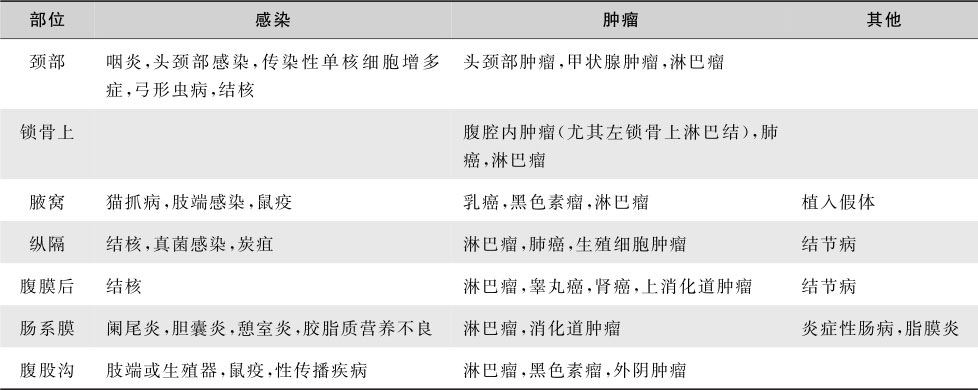
淋巴结触诊对浅表淋巴结肿大的鉴别甚有价值,可依据淋巴结肿大的部位、大小、数目、外形、硬度、压痛、活动度和局部皮肤表现进行综合分析。急性炎症性淋巴结最初肿大时多柔软、有压痛和痛感,表面光滑、无粘连,一般不引起巨大淋巴结肿大,应用抗生素治疗多能很快消退;慢性者稍硬,延时较久,但最后仍能缩小至消退。淋巴结结核引起淋巴结肿大多发生在颈血管周围,大小不一,有时成串,可互相粘连或与周围组织粘连,如发生干酪样坏死,亦可触到波动,晚期常溃破,不易愈合而形成瘘道,愈合后形成不规则瘢痕。破溃瘘道和瘢痕还见于放线菌病,但一般不出现在淋巴瘤和转移癌。恶性淋巴瘤引起的淋巴结肿大常为无痛性,质如硬橡皮,呈块状,可以融合,如生长很快的淋巴瘤淋巴结肿大也可有疼痛。恶性肿瘤转移的淋巴结肿大常质坚硬,固定,一般无压痛。通过体格检查还可从淋巴引流区域发现原发灶,不同部位淋巴结肿大需考虑的原发病不同(表16-5-18)。过度肿大的淋巴结可产生局部的压迫症状,如纵隔淋巴结肿大可引起咳嗽、肺不张、颈交感神经麻痹综合征、上腔静脉压迫征;肝门淋巴结肿大压迫总胆管引起黄疸;腹膜后淋巴结肿大可引起下肢、会阴部水肿。
表16-5-18 不同部位淋巴结肿大需考虑的原发病
(三)影像学检查
影像学检查包括常规平片、CT、超声、磁共振(MRI)和正电子发射计算机断层显像CT扫描(PET-CT)等,在纵隔和腹腔淋巴结肿大的诊断过程中尤为重要。对于腹腔和腹膜后淋巴结肿大,CT和超声检查有效,CT较为精确,超声检查则较经济、且患者无射线接触。MRI和PET对于淋巴结肿大并非首选,PET检查可用于淋巴瘤患者病变部位的检测,尤其对化疗后再评估有重要意义。
(四)淋巴结穿刺涂片及淋巴结印片
系通过细胞形态学检查来诊断淋巴结病变,其缺点是不能判断正常淋巴结结构的破坏,对分化好的淋巴细胞淋巴瘤难以鉴别,因此诊断淋巴瘤宜选用淋巴结活组织检查并且取材要完整的淋巴结组织,但淋巴结穿刺涂片对确定癌肿淋巴结转移具有重要价值。淋巴结穿刺涂片的优点是可以通过穿刺获得标本,不需要通过淋巴结活组织检查手术,方便、易于操作,且诊断速度大大加快。淋巴结穿刺活检,取其淋巴结穿刺和活组织检查两方面优点,但对淋巴瘤的病理诊断和分型仍不及完整淋巴结活组织检查。淋巴结印片细胞形态学检查还可以补充病理组织学检查的不足。因此,建议做淋巴结活组织检查的同时做印片行细胞形态学检查。
(五)淋巴结活组织检查
是诊断淋巴结病变的金标准,不仅可通过病理形态学诊断,还可以进行免疫组化和选择相应的单克隆抗体对淋巴细胞的免疫表型进行分析,对淋巴结病变的诊断有很大提高。对原因不明的深部淋巴结肿大,因需要进行CT引导下深部淋巴结穿刺或通过纵隔镜、腹腔镜或剖腹探查进行深部淋巴结活组织检查,具有一定风险,因此必须严格掌握指征。一般认为应先进行详细的影像学检查,特别是PET-CT疑有恶性可能,并且淋巴结>2~3cm,有多个淋巴结肿大,宜做侵入性检查。
【淋巴结肿大的诊治思路】
临床就诊的淋巴结肿大患者常因自行触摸或健康体检时发现头颈部、腋窝或腹股沟肿大淋巴结,亦或是在胸腹部影像学检查时发现。若肿大淋巴结较软、<2~3cm、无全身症状,可定期观察;若肿大淋巴结为多发、质地硬、>2~3cm,且伴有不明原因的发热、盗汗、体重减轻等症状时,需考虑淋巴结活检。头颈部淋巴结肿大活检前须仔细检查患者耳鼻和咽部,其他深部淋巴结活检可通过纵隔镜、腹腔镜、开腹或开胸手术进行。
患者可予血常规和外周血涂片检查随访,对传染性单核细胞增多症等感染患者和血液病患者有一定的诊断价值,若抗感染治疗后淋巴结缩小不明显、甚至持续增大者,仍需行淋巴结活检以明确诊断。
二、脾 大
无论任何体位,正常脾在肋弓下,一般是不能触及的。如能触到脾下缘即属脾大(splenomegaly)。但须注意确定脾大时宜除外脾下垂,后者亦可在肋缘下触及,见于内脏下垂患者或由左侧胸腔积液、积气致横膈下降所致,脾下垂者叩诊脾上界可降低。临床上判断脾大也可通过影像学诊断方法,正常脾脏前后径不超过10cm,厚度(宽)不超过6cm,上下径不超过15cm。临床上将脾大分为轻度、中度、高度肿大。肋缘下刚触及至肋下3cm以内属轻度肿大,3cm至脐水平位置为中度肿大,超过脐水平则为高度肿大或称巨脾。
【病因与发病机制】
(一)对脾脏功能需求过度所致的脾大
单核-巨噬细胞系统和免疫系统增生时都可能引起脾脏内细胞增殖而导致脾大。具体原因有:
1.感染性疾病
细菌、分枝杆菌、真菌、寄生虫、立克次体、病毒等感染都可刺激引起脾脏巨噬细胞与淋巴细胞增生,伴有充血而致脾大。伴脾大的急性感染性疾病包括传染性单核细胞增多症、病毒性肝炎、巨细胞病毒感染、伤寒与副伤寒、败血症、播散性结核、回归热、钩端螺旋体病及亚急性细菌性心内膜炎、AIDS、组织胞浆菌病、麻疹、疟疾等。伴脾大的慢性感染包括结核病、布鲁菌病、梅毒、真菌感染、锥虫病、慢性疟疾、黑热病、弓形虫病及血吸虫病等。直接侵犯脾的感染有脾脓肿、脾结核性肉芽肿等。
2.免疫调节功能异常
机体免疫调节功能异常时,脾内淋巴细胞、巨噬细胞增生引起脾大,可见于类风湿关节炎、系统性红斑狼疮、风湿热、白塞病、血清病、血管炎、药物热、甲状腺功能亢进症及白介素-2治疗等。类风湿关节炎合并白细胞减少及脾大的特殊类型称为Felty综合征。
3.单核-巨噬细胞系统增生
遗传性球形红细胞增多症等先天性红细胞膜缺陷性疾病、珠蛋白生成障碍性贫血、异常血红蛋白分子病及自身免疫性溶血性贫血等情况下,因为过量红细胞在脾内破坏,含铁血黄素在脾内沉积,脾脏单核-巨噬细胞增生,脾索增宽,血窦扩张而致脾大。
4.髓样化生
脾脏仅在胚胎期有造血功能,但在某些疾病或应急情况下脾脏可恢复造血功能,脾脏内各期造血细胞增殖且伴有巨噬细胞增生而导致脾大。如药物和放射线骨髓损害时、骨髓纤维化、真性红细胞增多症、原发性血小板增多症、白血病、肿瘤浸润和戈谢病也可发生不同程度的脾髓样化生而引起脾大。真性红细胞增多症脾大的原因尚与血容量增多有关。
(二)脾静脉或门静脉血流异常所致的脾大
脾静脉属门静脉系统,脾静脉压力增高、脾血回流受阻,可导致脾脏淤血和体积增大。充血性脾大常见于各种原因引起的慢性心功能不全、慢性缩窄性心包炎、肝静脉阻塞(Budd-Chiari综合征)、各种原因所致的肝硬化及门静脉或脾静脉血栓形成等,常伴腹水。由于红髓内单核-巨噬细胞增生,小梁增厚,脾索增宽,故充血性脾大易伴有脾功能亢进。
(三)脾内浸润所致的脾大
1.细胞内或细胞外物质沉积
溶酶体贮积症包括葡萄糖脑苷脂病(戈谢病)、神经鞘磷脂病(尼曼-匹克病)、海蓝组织细胞增生症等,系类脂质分解代谢途径中某种酶缺陷,使代谢中间产物在脾等器官大量堆积,组织细胞大量增生引起脾大,遗传性溶酶体贮积症常引起巨脾。淀粉样蛋白沉积于脾可引起脾大。
2.细胞浸润
许多血液系统恶性肿瘤,如白血病、淋巴瘤、Walderström巨球蛋白血症、血管免疫母细胞淋巴结病(免疫母细胞T细胞淋巴瘤)和恶性组织细胞病时恶性细胞易浸润脾并引起脾大。重度脾大多见于慢性白血病,慢性粒细胞白血病、慢性淋巴细胞白血病、幼淋巴细胞白血病、毛细胞白血病等。某些恶性淋巴瘤及恶性组织细胞病也可见巨脾。除黑色素瘤外,癌或肉瘤有脾转移者极其罕见。
Langerhans细胞组织细胞增生症常有脾大,在病变区有大量Langerhans细胞浸润,常与巨噬细胞、嗜酸性粒细胞、淋巴细胞形成炎性肉芽肿。噬血细胞综合征是一种单核-巨噬细胞系统良性反应性组织细胞增生症,52%的病例有脾大。此外,先天性囊肿和假性囊肿(脾血肿退行性变所致)、错构瘤、血管瘤、淋巴管瘤、纤维瘤等,都是某种非恶性的细胞成分在脾内浸润所致。
(四)其他
铍中毒、原发性脾大的原因不明。
【诊断与鉴别诊断】
(一)病史
详细询问病史对不明原因脾大的诊断和鉴别诊断具有重要价值。脾大伴全身症状如发热、盗汗、消瘦或淋巴结肿大者常由血液病、恶性肿瘤、感染和炎症性疾病引起。感染引起脾大常有特殊热型,如伤寒呈稽留热;疟疾、回归热呈间歇热;布鲁菌病为波状热;急性血吸虫病有间歇热或弛张热;亚急性感染性心内膜炎、结核病可呈不规则热或持续低热。肿瘤亦可引起长期不规则发热,恶性淋巴瘤有周期性发热。血液疾病脾大常伴有淋巴结肿大、血常规改变或网织红细胞增高。如脾进行性肿大常为血液恶性肿瘤。对慢性脾大,需询问有否病毒性肝炎病史、慢性疟疾病史、酗酒史、风湿性疾病以及疫水接触史,后者应考虑有血吸虫病的可能性。
(二)体格检查
体检时应注意脾的大小、质地、有否压痛及脾区摩擦感等。急性感染性脾大常为轻度肿大,质偏软,伴触痛,感染控制后脾可回复到正常大小。慢性感染所致脾大因有纤维组织增生常质地明显增加,感染控制后脾不回复到正常大小。直接侵犯脾的感染如脾脓肿、脾结核等常伴脾周围炎,脾触诊可有摩擦感伴压痛,听诊有摩擦音。巨脾因血供不足发生脾梗死者亦有明显的压痛、摩擦感和摩擦音。慢性充血性脾大外形规整,表面光滑;脾结核可扪及结节;淋巴瘤、脾囊肿和脾肿瘤可引起脾表面不平滑和变形。
(三)实验室检查
血常规、血涂片、网织红细胞计数、厚血片查疟原虫、肝肾功能、血红蛋白电泳、血清蛋白电泳、自身抗体检查、结核酶联免疫斑点试验(T-spot)、肝炎病毒检查、LDH和抗人球蛋白试验等在脾大的鉴别诊断中是常用的检查。血常规和骨髓象检查对分析原因不明的脾大有重要参考价值。脾大伴中性粒细胞增多大多提示有细菌感染,伴淋巴细胞增多并有异形淋巴细胞出现提示为病毒感染。若周围血液出现大量幼稚细胞应考虑为白血病,老年患者脾大伴成熟淋巴细胞增多要考虑慢性淋巴细胞白血病。脾大伴红细胞或血小板增多,应考虑有骨髓增生性肿瘤的可能。脾大伴贫血、网织红细胞增多和黄疸应考虑溶血性贫血。
(四)影像学检查
影像学检查在脾大的诊断中主要用来测定脾大小、估计脾的内部结构、其他脏器评估和引导穿刺。超声检查是最常用的方法,没有电离辐射,可以精确提供脾脏的大小,便于重复。CT可以提供连续的成像,有助于鉴别脾源性肿瘤和脓肿。正电子发射计算机断层显像CT扫描(PET-CT)在确定脾占位病变的良恶性方面有一定参考价值。核素 51 Cr或 59 Fe 标记红细胞注入体内测定脾区放射量,能协助了解脾功能有无异常及寻找有无副脾的存在。 99m 锝肝脾显像尚能帮助肝硬化致脾大的诊断。
(五)活组织检查
脾大的原因不明,通过上述步骤仍不明原因者,可进行脾活组织检查以明确诊断。鉴于脾所处的部位和易出血的特征,脾穿刺术要慎重考虑,所谓的脾活组织检查即指脾切除后活检。脾切除可经剖腹手术或腹腔镜下进行,但腹腔镜下手术时组织易损坏,可能减低切脾后病理确诊的价值。有些患者有副脾或切脾时脾细胞腹腔内种植,导致切脾治疗的效果不明显,可以外周血红细胞中是否含有豪-胶小体以鉴别。
【脾大与脾功能亢进】
脾大约占健康青年的3%和所有住院患者的5%。1907年Chauffard开始提出脾功能亢进的概念,但迄今为止,脾功能亢进的诊断尚无法精确定义。脾功能亢进症(hypersplenism)简称脾亢,是一种综合征而不是一个独立的疾病,临床表现为脾大、一种或多种血细胞减少、骨髓造血细胞代偿性增生、脾切除后血象可恢复。
脾亢主要分为原发性和继发性两大类。原发性脾亢是指原发性脾增生,无其他明确的可以导致脾大的原发病因,可见于非热带性特发性脾大、原发性脾性粒细胞减少、原发性脾性全血细胞减少、脾性贫血或脾性血小板减少症。继发性脾亢多有明确的导致脾大的因素,常由门静脉高压和脾内浸润引起,可见于感染伴脾大、充血性脾大、炎症性肉芽肿、恶性肿瘤、慢性溶血性疾病、溶酶体贮积症、骨髓增殖性肿瘤等。随着对脾大病因诊断手段的日益改进,原发性脾亢的比例已逐渐减少。
脾功能亢进引起血细胞减少的机制,可能跟过分阻留、过分筛选和吞噬作用有关。正常人脾内并无红细胞或白细胞贮藏作用,但约1/3的血小板及部分淋巴细胞却被阻留在脾。当脾有病理性显著肿大时,不但更多血小板(50%~90%)及淋巴细胞在脾内阻留,而且也可有30%以上的红细胞在脾内滞留,导致周围血中血小板及红细胞减少。此外,脾亢时脾内单核-巨噬细胞系统过度活跃,而脾索内异常红细胞(如球形红细胞及受抗体、氧化剂或其他化学毒物、物理因素损伤的红细胞等)明显增多并为巨噬细胞清除,导致周围血中红细胞明显减少。有些红细胞膜上出现海因小体,或浆内有豪-胶小体,甚至疟原虫的滋养体,当自脾索进入血窦时,红细胞因其海因小体或豪-胶小体被夹在窦壁基膜小孔而进退两难,最后为窦壁巨噬细胞挖除,同时红细胞膜受损。反复多次受损后红细胞成为球形红细胞,终至无法通过基膜小孔而被吞噬。此外,尚有学者提出脾亢时脾产生过多的体液物质,以抑制骨髓造血细胞的释放和成熟;也有认为脾亢是一种自身免疫性疾病,但均缺乏有力的佐证,有待研究证实。
【脾大的诊治思路】
脾大患者的主诉不一,可以左上腹痛、左季肋区不适或进食后腹胀等就诊,左上腹痛且向左肩放射提示脾梗死,脾破裂罕见。有些患者的脾大在常规体检或其他原因行影像学检查时发现,亦可由于不明原因的全血细胞减少就诊而发现。脾大的原发病多为全身性疾病,如传染性单核细胞增多症、白血病和淋巴瘤、类风湿关节炎、结节病、肝硬化、疟疾等。确诊并治疗后,需随访脾脏大小,若治疗成功,则脾脏可缩回正常大小。
诊断脾功能亢进有赖于以下各项指标,①脾大;②一系或多系血细胞减少,一般早期病例只有白细胞或血小板减少,晚期病例发生全血细胞减少;③代偿性增生的骨髓像,部分病例还可同时出现成熟障碍;④脾切除后血细胞数接近或恢复正常。在考虑脾亢的诊断时,以前三条尤为重要。确诊原发性脾亢需严格除外可以引起脾大的其他病因后可尝试肾上腺皮质激素,对血细胞减少有一定效果。
无明确主诉的脾大患者确诊较困难,可能为隐匿的肝病或自身免疫性疾病所致,需仔细随访,对隐匿性肝病、门脉高压者,不需要选择切脾。其他原因的脾大有明显压迫症状、严重血细胞减少且有相应症状(反复感染、出血等),可考虑行脾切除。若怀疑肿瘤所致的脾大,患者常伴有发热、盗汗、体重减轻、影像学检查发现局灶性病变,亦可作为切脾的指征。切脾患者应进行充分的术前准备若无上述的症状和检查结果,则需密切随访和重复检查,切脾应慎重。
主要参考文献
1.Goldman L,Schafer AI.Goldman's Cecil Medincine.25th ed.Philadelphia:Elsevier Saunders,2016:1138-1141.
2.Greer JP,Foerster J,Rodgers GM,et al.Wintrobe's Clini-cal Hematology.12th ed.Philadelphia:Lippincott Williams&Wilkins,2009:1637-1654.

