第七章
止血与血栓
第一节
出血性疾病概述

王伟光 程韵枫
出血性疾病指由于遗传性或获得性原因,导致患者止血、凝血及纤维蛋白溶解机制的缺陷或抗凝机制异常所致的一组疾病。该类疾病有以下特点:①自发性或轻微外伤出血难止;②出血常发生于多部位或非寻常部位,呈广泛性或局部性;③病情反复发作,且持续时间较长;④不能解释的手术或创伤时出血严重;⑤一般止血药物效果差,血液制品效果佳;⑥部分患者有明显的出血史或家族史。该组疾病病因复杂,在临床上占血液疾病的30%,近年来随着分子生物学、免疫学和生物化学的发展,对其有更新的认识。
【正常止凝血机制】
正常的止血由三个过程组成。初级止血过程(一期止血)依赖于血小板的激活,形成血小板栓子(白色血栓);次级止血过程(二期止血)依赖于凝血机制参与,形成纤维蛋白凝块(红色血栓);纤维蛋白溶解过程在于清除纤维蛋白,恢复正常的血流。
(一)血管壁的止血功能
参与止血作用的血管主要有小动脉、小静脉和毛细血管,其基本结构可分为内膜层、中膜层和外膜层。
1.血管壁的结构
内膜层由内皮细胞组成,血管内皮细胞是位于循环血液与血管壁内皮下组织之间的单层细胞,内皮细胞有一种特异的细胞器被称为棒杆状小体或Weibel-Palade小体,其为血管性血友病因子(von Willebrand Factor,v WF)贮存加工的场所。内皮细胞合成和分泌的其他分子包括血小板活化因子(platelet activating factor,PAF),凝血酶敏感蛋白(thrombospondin,TSP),纤维联接蛋白(fibrinectin,Fn),纤溶酶原激活物抑制剂-1(plasminogen activator inhibitor-1,PAI-1),凝血酶调节蛋白(thrombomodulin,TM)和层素(laminin,Ln)。vWF参与血小板与血管基底膜的黏附,内皮细胞表面的糖萼(glycocalyx)蛋白是多种受体所在部位。
血管内皮细胞结构和功能完整时,血小板对管壁是排斥而被动进入循环的,前列环素和一氧化氮作为强烈的血管扩张剂在局部起到了对血小板的抑制作用。但在血管损伤处,这些活性物质减少,为血小板的黏附聚集提供了基础。
中膜层是介于内皮细胞和外膜层之间的结构,包括基底膜,微纤维,Ⅲ、Ⅳ、Ⅴ型胶原,平滑肌和弹力纤维。基底膜是一种胶原蛋白,能支撑内皮细胞及诱导血小板的黏附和聚集,并启动内外源性凝血过程;平滑肌和弹力纤维参与血管的收缩功能。
血管内皮细胞和中膜层均可表达组织因子(tissue factor,TF),启动外源性凝血过程。
外膜层由结缔组织构成,是血管与组织之间的分界层。
2.血管壁的止血功能
(1)增强收缩反应:
血管损伤时通过神经轴突反射和缩血管活性物质如内皮素-1(endothelin-1,ET-1)、儿茶酚胺、血管紧张素、血栓烷A 2 (thromboxane A 2 ,TXA 2 )及5-羟色胺(5-HT)的释放使受损血管收缩以利于止血。
(2)激活血小板:
主要通过下述三个途径实现。vWF的作用;PAF,迄今所知最强的血小板诱聚剂;TXA 2 ,除收缩血管外,尚能促进血小板黏附、聚集和活化。在上述因子的作用下,以血管内皮细胞下胶原的暴露为基础,使血小板发生黏附和聚集,形成血小板栓子。
(3)激活凝血系统:
胶原的暴露激活凝血因子Ⅻ、血管内皮细胞表达Ⅺ/Ⅺa活性,并结合活化的Ⅹa加速内皮细胞表面凝血酶原的激活;内皮细胞表达TF;分别启动内、外源性凝血过程。
(4)抑制纤溶:
通过血管内皮细胞合成分泌PAI-1实现。
(5)增高局部血黏度:
通过激活凝血因子Ⅻ和激肽释放酶原,生成激肽;激活血小板释放血管通透性因子;两者的作用使血管通透性增加、血浆外渗、血液浓缩、血黏度增高,血流缓慢,有利于止血。
(二)血小板的止血功能
1.血小板结构和生化组成
电镜下,血小板由表面结构、骨架、细胞器和特殊膜系统四部分组成,各部分生化组成各有特点。
(1)血小板表面:主要由糖萼蛋白和细胞膜组成。糖萼蛋白为细胞外衣,由糖蛋白和糖链部分组成,是许多血小板膜受体(ADP、肾上腺素、胶原、凝血酶)的所在部位;细胞膜主要由蛋白质和脂质组成,其中膜脂质磷脂酰丝氨酸主要分布在内侧面,在血小板激活时转向外侧,可能为血小板第3因子(platelet factor 3,PF 3 )的主要成分;膜蛋白主要为糖蛋白,其中GPⅠb/Ⅸ参与血小板的黏附,GPⅡb/Ⅲa与血小板聚集有关,GPⅠa/Ⅱa是胶原的受体,GPⅠc/Ⅱa为Fn的受体,GPⅣ是TSP的受体,GPⅤ参与血小板的黏附。
(2)骨架系统:包括微管和微丝,前者维持血小板的形状,后者的肌动蛋白细丝和肌球蛋白粗丝构成血小板的收缩蛋白,参与了血小板的收缩、伪足形成和释放反应。
(3)电镜下血小板内有许多细胞器,其中重要的是α颗粒、致密(δ)颗粒、溶酶体(γ)颗粒。
1)α颗粒:主要含以下活性物质:①β-血小板球蛋白(β-thromboglobulin,β-TG),是血小板特异的蛋白质,可抑制血管内皮细胞产生PGI 2 ,促进血小板的聚集。②血小板第4因子(platelet factor 4,PF 4 ),中和肝素的抗凝活性,减慢凝血酶的灭活,促进血栓形成。③TSP,是血小板α颗粒的主要糖蛋白,但非血小板特异的蛋白质,主要通过依赖和非依赖血小板纤维蛋白原受体系统的两种机制促进血小板的聚集。④血小板源生长因子(platelet derived growth factor,PDGF),在凝血酶作用下释放,刺激成纤维细胞和肌细胞生长和分裂,参与动脉粥样硬化的发生。⑤Fn,非血小板特异,在血小板受凝血酶或胶原刺激后释放于膜表面,介导血小板对胶原的黏附反应。
2)δ颗粒:含有①ATP和ADP,前者维持血小板形态、功能和代谢活动,后者促进血小板的聚集和释放;②5-HT,接受凝血酶的刺激,促进血小板的聚集和血管收缩。
3)γ颗粒:含有多种酸性水解酶和组织蛋白酶,是血小板的消化结构。
(4)血小板的特殊膜系统:主要包含开放管道系统(open canalicular system,OCS)和致密管道系统(dense tubular system,DTS)。OCS是血小板内与血浆物质交换的通道,DTS参与花生四烯酸代谢、前列腺素的合成、血小板的收缩活动和释放反应。
2.血小板的止血功能
(1)黏附功能:
血小板黏附(platelet adhesion)是指血小板黏着于血管内皮下组分和其他异物表面的功能,血小板膜糖蛋白GPⅠb-Ⅸ作为vWF受体,通过vWF和内皮下胶原结合来实现。
(2)聚集功能:
黏附的血小板发生“释放反应”,释放出储存颗粒组分如二磷酸腺苷(ADP),并通过环氧化物酶反应由花生四烯酸合成血栓烷A 2 。ADP、TXA 2 和其他因子共同作用,活化更多的血小板。激活的血小板在Ca 2+ 存在的条件下,膜糖蛋白GPⅡb-Ⅲa作为纤维蛋白原受体,通过纤维蛋白原,vWF,Fn形成血小板聚集(platelet aggregation)。
(3)促凝功能:
指血小板参与凝血的过程。PF 3 促凝活性,特指PF 3 参与凝血因子Ⅸa-Ⅷa-Ca 2+ 和Ⅹa-Ⅴa-Ca 2+ 复合物的形成;接触产物生成活性(contact product-forming activity,CFPA),血小板受ADP或胶原刺激后,CFPA从血小板膜磷脂成分释放,激活因子Ⅻ;胶原诱导的凝血活性(collagen induced coagulant activity,CICA),血小板受ADP或胶原刺激后,CICA从血小板膜磷脂成分释放,激活因子Ⅺ;α颗粒中凝血因子(Ⅴ、Ⅺ、纤维蛋白原)的释放。
(4)释放反应:
指血小板在活化中由于病理因素或诱导剂作用将其颗粒内容物通过OCS释放到细胞外的过程。β-TG、PF 4 、血栓烷素 B 2 (TXB 2 )、血小板颗粒膜蛋白-140(GMP-140)已作为检测血小板活化分子标志物。
(5)血块收缩功能:
通过激活血小板的肌动蛋白细丝和肌球蛋白粗丝的相互作用,形成的血小板伪足向心性收缩来完成。
(6)维护血管内皮的完整性:
血小板填充血管内皮细胞脱落形成的孔隙,参与血管内皮细胞的再生和修复,增加血管壁的抗力,减低血管壁的通透性和脆性。
(三)血液凝固
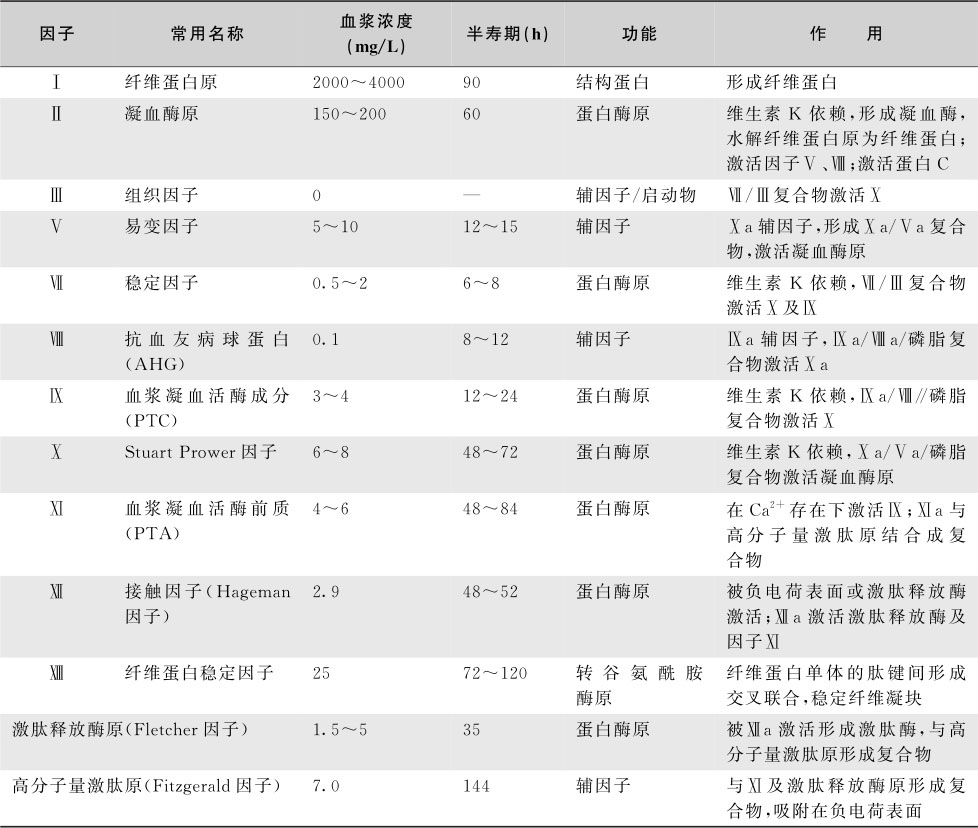
1.凝血因子的特性
凝血因子迄今已知至少有14种,包括经典凝血因子12个和激肽系统2个,经典凝血因子采用罗马数字命名。除Ⅳ因子是钙离子外,其余均为蛋白质;除Ⅲ因子存在于组织外,其余均存在于血浆中。凝血因子的特性见表16-7-1。
表16-7-1 各种凝血因子的特征
2.凝血过程的瀑布学说
凝血反应是凝血因子通过酶促反应而相继被激活,以瀑布效应形成纤维蛋白的过程。
(1)内源性凝血途径:
指从FⅫ被激活到FⅩa形成的过程。血浆中FⅫ以酶原形式存在,带负电荷的异物表面或损伤的血管内皮下成分与之接触,即可激活FⅫ成为FⅫa(接触激活);FⅫa将前激肽释放酶(prekallikrein,PK)激活为激肽释放酶(kallikrein,KK),KK可反馈地通过酶解方式激活FⅫ(液相激活)。FⅫa激活FⅪ成为FⅪa,在此过程中,高分子量激肽原(high molecular weight kininogen,HMWK)发挥辅因子作用。FⅪa在Ca 2+ 存在下,激活FⅨ为FⅨa。FⅨa、FⅧa和Ca 2+ 在活化的血小板提供PF 3 作用下形成FⅩ激活复合物,该复合物激活FⅩ成为FⅩa。
(2)外源性凝血途径:
血管内皮细胞受损或组织损伤,均能将TF释放入血;或者作为一种跨膜蛋白,TF表达于血管内皮细胞表面。TF作为辅因子与FⅦ结合形成FⅦ/TF复合物,该复合物被血液中痕量FⅩa激活形成FⅦa/TF,后者正反馈激活FⅩ形成FⅩa。
(3)共同通路:
FⅩa在FⅤa和Ca
2+
存在条件下,以活化血小板提供的PF
3
为磷脂表面,形成凝血酶原酶复合物,该复合物由FⅩa首先水解凝血酶原(FⅡ)分子精氨酸271-苏氨酸272肽键,释放出源于分子氨基端的片段,即凝血酶原碎片1+2(F
1+2
)后,形成尚不具备酶解活性的前凝血酶2(prothrombase 2);前凝血酶2仍为单链分子,再经FⅩa进一步作用于精氨酸320-异亮氨酸321肽键,形成含有A、B两条多肽链的凝血酶(FⅡa,thrombin)。FⅡa先后裂解纤维蛋白原2条Aα链的氨基端精氨酸16-甘氨酸17肽键和2条Bβ链的氨基端精氨酸14-甘氨酸15肽键,相继脱下含16个氨基酸残基的纤维蛋白肽A与含14个氨基酸残基的纤维蛋白肽B(FPA,FPB)后形成纤维蛋白单体,单体间通过自动聚合形成可溶性纤维蛋白单体复合物,该复合物在F
 a作用下形成交联,产生非可溶性纤维蛋白,完成血液凝固过程。F
1+2
、FPA、FPB作为体内凝血激活状态分子标志物,而用于对机体凝血过程激活的预测。
a作用下形成交联,产生非可溶性纤维蛋白,完成血液凝固过程。F
1+2
、FPA、FPB作为体内凝血激活状态分子标志物,而用于对机体凝血过程激活的预测。
3.经典凝血途径的修正
经典的瀑布学说认为接触激活相是内源性途径的始动阶段,内源性途径是生理性止凝血过程的主要途径,外源性途径是次要的或辅助性的。
研究表明:内源性凝血过程中的接触激活不参与生理性止血,而主要与病理条件下(如内毒素血症致DIC)血压降低和炎症反应有关。因为KK作用于HMWK使之生成缓激肽,缓激肽通过刺激血管内皮细胞释放内皮衍生松弛因子(endothelial derived relaxing factor,EDRF)与PGI 2 引起血管扩张,说明接触激活是DIC引起低血压休克的基本原因;其次FⅫa与KK对白细胞具有趋化效应,且能激活补体系统,故与炎症反应发生发展有关。另有研究发现,FⅦ缺陷可导致严重出血倾向;补充FⅦ或应用组织因子通路抑制物(tissue factor pathway inhibitor,TFPI)单克隆抗体可以纠正血友病A(FⅧ缺陷)的出血倾向。因此Davie与Broze对经典的瀑布学说进行了修正,认为凝血过程分为两个阶段,首先是启动阶段,确认生理性止血过程是由组织因子启动,由于TFPI的作用,只可能形成少量凝血酶,不足于完成凝血过程;然后是放大阶段,即少量凝血酶反馈激活血小板与FⅪ,激活内源性凝血过程。在病理性止血过程中凝血过程是由于内皮细胞或单核细胞在损伤、感染、内毒素、细胞因子、缺氧作用下表达产生TF而启动的,组织因子在凝血过程中属于主角地位。目前一般认为,外源性凝血途径在体内生理性凝血反应的启动中起关键性作用,组织因子被认为是生理性凝血反应的启动物,而内源性凝血途径对凝血反应开始后的维持巩固阶段可能发挥作用(图16-7-1)。
(四)抗凝系统的作用
体内凝血的启动及凝血因子的活化,同时引起凝血抑制物的干预。体内抗凝系统大致可分为两个方面:细胞抗凝机制和体液抗凝机制。前者指单核-巨噬细胞系统对激活的凝血因子、凝血酶原酶复合物及可溶性纤维蛋白单体的吞噬作用,而后者主要包括以下方面。
1.抗凝血酶系统
曾称为抗凝血酶-Ⅲ和肝素辅因子Ⅰ,是最重要的抗凝因子,主要作用机制是其羧基端一个精氨酸残基与丝氨酸蛋白酶活性部位丝氨酸相结合,从而形成一个1∶1不可逆的共价复合物,主要灭活凝血酶,尚能抑制FⅩa、Ⅸa、Ⅺa、Ⅻa。肝素主要作用于抗凝血酶的赖氨酸残基而放大其抗凝血酶活性。
2.蛋白C系统
主要包括蛋白C(PC)、蛋白S(PS)、凝血酶调节蛋白(TM)和蛋白C抑制物。PC在凝血酶作用下形成活化的蛋白C(APC),主要能灭活凝血辅因子FⅤa、FⅧa;阻碍因子Ⅹa与血小板结合;促进纤维蛋白溶解;PS主要通过加速APC对FⅤa的灭活而发挥作用;TM固定于胞膜上,本质是凝血酶受体,在Ca 2+ 存在的条件下,加速PC的活化。
3.TFPI系统
直接抑制FⅩa,并以依赖FⅩa形成在Ca 2+ 存在的条件下,抑制TF/FⅦa复合物。
4.其他
包括肝素辅因子Ⅱ、肝素、α 2 -巨球蛋白、α 1 -抗胰蛋白酶等。
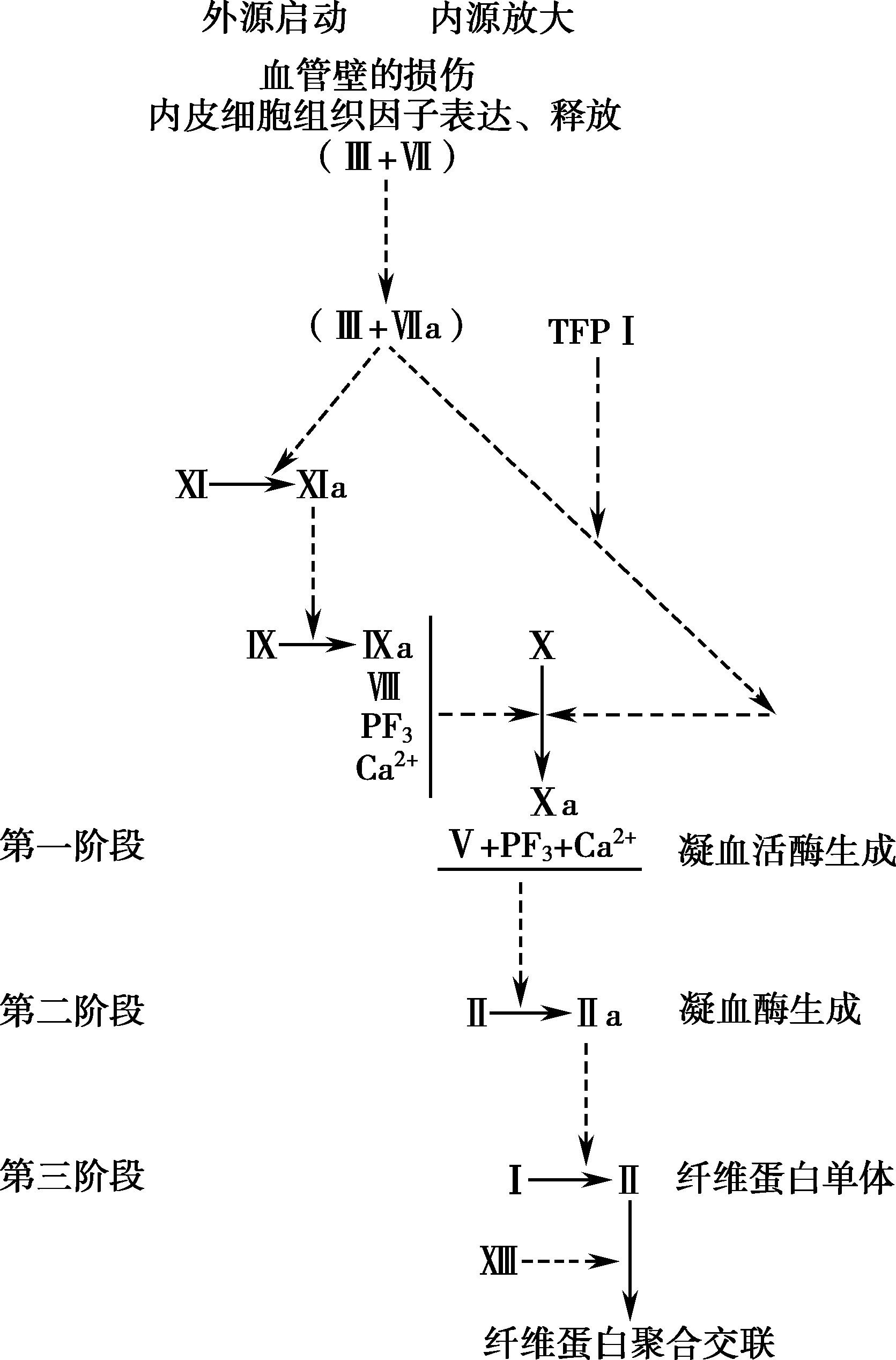
图16-7-1 凝血过程瀑布学说
作用:
 ;生成:
;生成:
 抑制:
抑制:

(五)纤维蛋白溶解系统
纤溶系统主要由纤溶酶原和纤溶酶、纤溶酶原激活剂、纤溶抑制物组成(表16-7-2);机体对纤维蛋白的清除主要依靠纤溶酶对纤维蛋白的降解。纤溶系统的激活主要包括:①内激活途径:凝血接触激活中产生FⅫa及FⅫa碎片(Ⅻf)激活PK形成KK,KK激活纤溶酶原成纤溶酶。②外激活途径:血管内皮细胞在各种病理因素作用下释放t-PA从而激活纤溶酶原,此过程受到纤溶酶原活化剂抑制物(PAI-1)调节。③外源性激活途径:将体外的激活纤溶系统的制剂SK、UK、rt-PA注入体内激活纤溶系统达到溶栓目的。原发性纤溶亢进主要由外激活途径完成,而继发性纤溶亢进由内、外两条激活途径实现。
纤维蛋白(原)降解产物(FDP)见图16-7-2。
【分类】
出血性疾病分遗传性和获得性两大类,并以病理环节为基础分成以下类型。
(一)血管壁异常
因血管壁结构及其周围支撑组织功能异常或受损所致。遗传性临床少见,如遗传性毛细血管扩张症、巨大海绵状血管瘤、马方综合征等。获得性包括免疫性(过敏性)、感染性(败血症细菌栓塞性)、化学性、代谢性(类固醇性)及机械性紫癜。
表16-7-2 纤溶系统的组成成分及作用
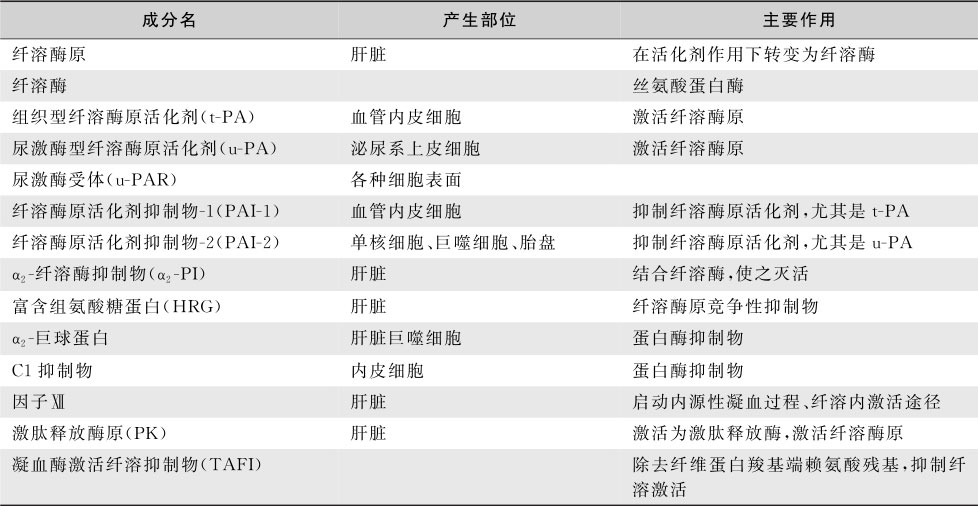
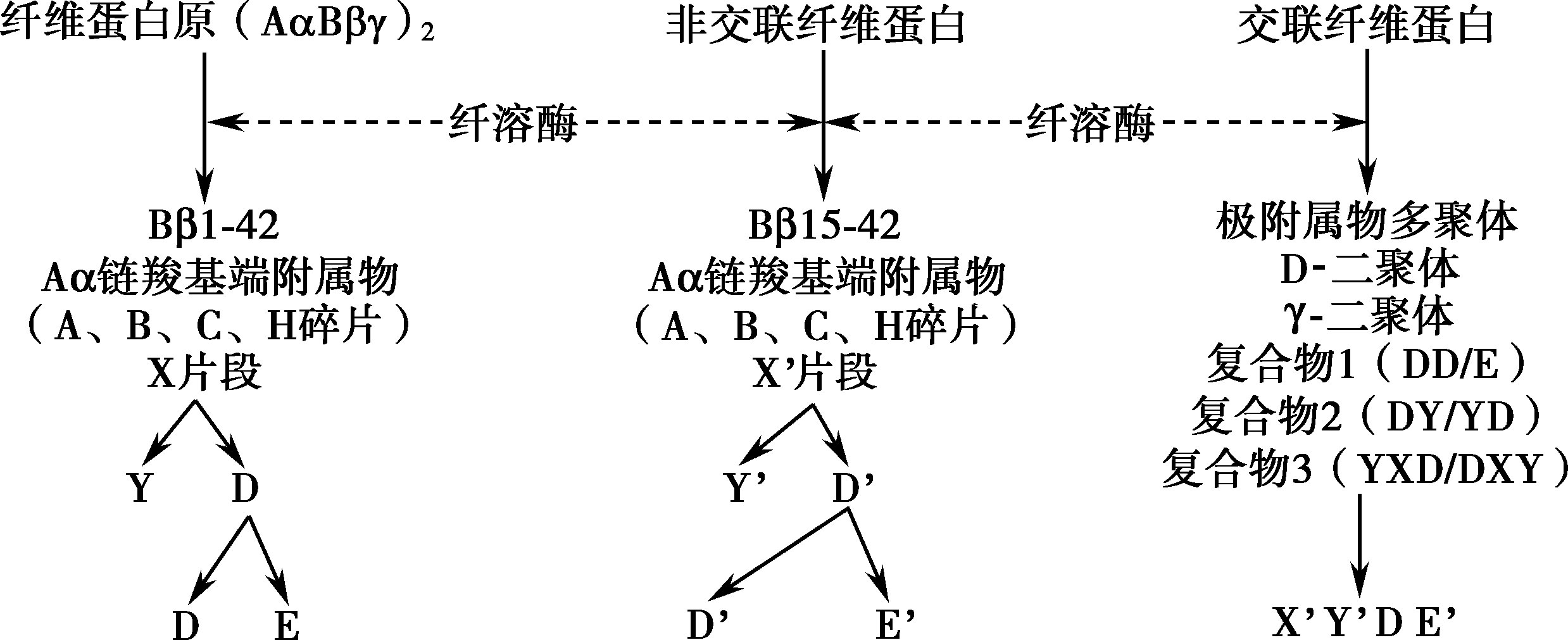
图16-7-2 纤维蛋白(原)降解产物
(二)血小板异常
1.血小板数量减少
①生成减少,指各种原因导致巨核细胞(血小板)生成障碍,如再生障碍性贫血、急性白血病、肿瘤骨髓浸润等。②消耗过多,如弥散性血管内凝血(DIC)、血栓性血小板减少性紫癜(TTP)、溶血尿毒综合征(HUS)等。③破坏过多,如免疫性血小板减少性症(ITP)。
2.血小板增多症
常伴有血小板功能下降,见于反应性血小板增多症及骨髓增殖性疾病。
3.血小板功能缺陷
①先天性:a.黏附异常(巨血小板综合征、血管性血友病等);b.分泌异常(灰色血小板综合征、贮存池病等);c.活化异常(环氧化酶缺乏症、TXA 2 合成酶缺乏症等);d.聚集异常(血小板无力症等);e.促凝功能缺陷(PF 3 缺乏症)。②获得性:药物、尿毒症、免疫性疾病、肝病、白血病、骨髓增生异常综合征、骨髓增殖性肿瘤、异常蛋白血症及抗血小板抗体等。
(三)凝血因子异常
临床上以获得性因素为多见,主要包括:①重症肝病:缺乏纤维蛋白原,凝血酶原,因子Ⅴ、Ⅶ、Ⅸ、Ⅹ、Ⅺ、Ⅻ、
 等。②维生素K依赖性因子Ⅱ、Ⅶ、Ⅸ、Ⅹ缺乏,见于胆道疾病、广谱抗生素长期应用,口服抗凝剂等。
等。②维生素K依赖性因子Ⅱ、Ⅶ、Ⅸ、Ⅹ缺乏,见于胆道疾病、广谱抗生素长期应用,口服抗凝剂等。
遗传性因素最多见的是血友病A(因子Ⅷ缺乏);其次血友病B(因子Ⅸ缺乏),遗传性凝血因子Ⅺ缺乏症,纤维蛋白原和凝血酶原疾病,包括活性下降和抗原性异常,因子Ⅴ、Ⅶ、Ⅹ、Ⅻ、
 、激肽释放酶原及高分子量激肽原缺乏症,家族性复合性凝血因子缺乏症等。
、激肽释放酶原及高分子量激肽原缺乏症,家族性复合性凝血因子缺乏症等。
(四)纤维蛋白(原)溶解亢进
获得性包括原发性和继发性两种。所谓原发性者指组织型纤溶酶原激活物(t-PA)或尿激酶型纤溶酶原激活物(u-PA)释放入血(前列腺、甲状腺、胰腺手术过度挤压)或抗纤溶酶活性降低(肝病、肿瘤)所致纤溶亢进;继发性指凝血反应启动后因子Ⅻa激活激肽释放酶原生成激肽释放酶,后者激活纤溶系统,同时纤维蛋白沉积于血管内皮细胞表面导致t-PA的释放,见于DIC及各种血栓性疾病。
先天性少见,包括α 2 -纤溶酶抑制物(α 2 -PI)缺乏症、纤溶酶原活化物抑制物(PAI)缺乏症等。
(五)病理性抗凝物质过多
1.因子Ⅷ抑制物 血友病性指反复输注因子Ⅷ产生抗体后致病;非血友病性见于妊娠、自身免疫性疾病、淋巴系统恶性增殖性疾病、药物反应等。原发性多见于50岁以上患者,常无明显诱因。
2.获得性因子Ⅸ、Ⅺ、Ⅴ、
 抑制物。
抑制物。
3.狼疮样抗凝物质 本质为抗磷脂抗体,体外有抗凝血活性,体内损伤血管内皮细胞,导致血栓形成。
4.组织因子抑制物。
5.高肝素血症 见于肝素应用过量,重症肝病(肝素灭活能力下降)。
(六)复合因素引起的出血性疾病
临床上较常见的为各种致病因素导致的弥散性血管内凝血和重症肝病引起的出血。
【临床表现】
(一)病史特征
1.年龄
①出生后出现的出血如脐带断端出血、幼年期出血是遗传性疾病的特征,见于各种凝血因子的缺乏;产后数小时出现的紫癜和瘀斑伴血小板减少,应考虑同种免疫性血小板减少性紫癜;②年轻或成年后出血多为获得性因素所致,如免疫性血小板减少症、凝血因子抑制物出现。轻度血友病可在成年后发病;③老年期出血常与血管病变有关;④随年龄增加而改善的出血是血管性血友病及先天性血小板功能缺陷的临床特征;⑤老年人的免疫性血小板减少应警惕继发于淋巴系统恶性增殖性疾病的可能,凝血因子活性下降应考虑循环中存在病理性抗凝物质。
2.性别
①血管性血友病属于常染色体遗传,男女均可患病;②血友病A在男性中占绝大多数,女性甚为罕见;③育龄期女性免疫性血小板减少症发病率高于男性。
3.手术和创伤
①无诱因的出血或临床上原发病不能解释的出血,常提示患者有严重的出血性疾病,如:重症血友病或DIC;②小手术或轻度外伤后出血,特别是渗血不止是遗传性疾病的重要特征;③小手术或轻度外伤后延迟出血见于凝血机制障碍,而不常见于血管-血小板性出血;④大型手术,特别是联合器官移植、严重创伤后的出血如果排除大血管损伤表明可能伴发DIC;⑤子宫、卵巢、前列腺、胰腺、甲状腺是t-PA含量最丰富的部位,这些部位手术后出血应考虑原发性纤维蛋白原溶解亢进的可能;⑥小伤口或注射部位出血不止常提示有血小板减少、严重凝血机制缺陷或复合性止凝血机制紊乱。
4.药物
多种药物常通过以下几个方面与出血性疾病相关:①药物过敏性紫癜,相关药物如:青霉素、链霉素、磺胺药、异烟肼等。②药物免疫性血小板减少性紫癜,相关药物包括:水杨酸类解热镇痛药、多种抗生素、植物碱类如奎尼丁、镇静催眠药、磺胺衍化物、氢氯噻嗪、洋地黄类、金盐、西咪替丁等。③药物致血小板生成减少,如抗肿瘤药物引起骨髓抑制;氢氯噻嗪、雌激素类选择性抑制巨核细胞生长。④影响血小板功能药物,包括:影响前列腺素合成药物如阿司匹林,增加血小板cAMP类药物如双嘧达莫,肝素及纤溶剂,β-内酰胺类抗生素,低或中分子右旋糖酐。⑤广谱抗生素导致肠道菌群失调,维生素K合成减少。⑥诱导凝血因子抗体形成,如:青霉素、链霉素、磺胺药、异烟肼等。⑦医源性抗凝、溶栓药物应用过量。值得注意的是,青霉素可通过过敏性紫癜、免疫性血小板减少、诱导凝血因子抗体产生多种机制引起出血倾向。
5.妊娠、分娩
①妊娠常合并免疫性血小板减少,分娩后可缓解;②易并发因子Ⅷ抑制物的产生;③并发血栓性微血管病如:TTP、HUS、HELLP(妊娠-肝酶升高-溶血-血小板减少)综合征、急性脂肪肝;④产科意外(羊水栓塞、胎盘早剥、前置胎盘、宫内死胎、感染性流产)可导致DIC。
6.家族史
与出血性疾病相关遗传方式有:①常染色体显性遗传;②常染色体不完全显性遗传;③常染色体隐性遗传;④X性连锁隐性遗传。
(二)出血频度和程度
1.经常性严重出血
提示有遗传性凝血机制障碍、重症血管性血友病、重症肝病。
2.间歇性反复出血
为血小板减少性疾病的常见表现。固定部位的反复出血表明局部血管性病变如遗传性毛细血管扩张症。
3.一过性出血
通常提示获得性疾病如:病毒感染导致急性血小板减少性症;凝血因子抑制物的形成;药物免疫性血小板减少症及过敏因素等。
4.暴发性出血
多为严重血小板减少及复合性止凝血障碍所致。见于:①急性血小板减少症:病因包括急性ITP、TTP、药物免疫性血小板减少、血小板GPⅡb/Ⅲa拮抗药相关性血小板减少;②暴发性紫癜:如 Waterhouse-Friderichsen 综合征,主要表现以紫癜为首发症状的严重感染,伴血小板进行性下降,酸中毒、休克和神志改变;③DIC暴发期;④传染病如流行性出血热、钩端螺旋体病等。
5.延迟出血
是凝血机制障碍性疾病的特征,由于缺乏牢固纤维蛋白网支持,血小板血栓增大后崩解,而随后发生出血。
(三)出血倾向的特征
1.皮肤黏膜出血
是血小板和血管性出血性疾病最常见最易发现的症状和体征,可表现为:①出血点、紫癜和瘀斑:大片瘀斑是严重血小板减少、凝血障碍性疾病的特征。②血疱:为大小不等的口腔及舌部位的黏膜下出血,常见于暴发性紫癜、急性血小板减少症等疾病。③鼻出血:局部异常可为遗传性毛细血管扩张症,全身性疾病多为血小板减少。④齿龈出血:多有局部炎症引起,严重者见于血小板减少及凝血功能障碍。
2.深部器官出血
①血肿:为深部皮下、肌肉及其他软组织出血的表现,多见于凝血机制障碍,轻度外伤后或自发性血肿为血友病的特征。②关节积血:多见于负重的关节,尤其是膝关节,多见于严重凝血机制障碍,如血友病。③浆膜腔出血:血性浆膜腔积液排除外伤及肿瘤性因素,多见于凝血机制障碍。④眼底出血:见于重症血小板减少。
3.内脏出血
①呼吸道:表现为咯血。如果除外呼吸道病变,见于重症血小板减少和凝血机制障碍。另外,肺脏组织富含t-PA,纤溶亢进参与出血过程。②消化道:如果排除消化道本身病变,凝血因子缺陷和血小板减少可能为消化道出血的原因,但复合因素更为多见。③泌尿道:肿瘤性疾病出现血尿,如果排除转移,应考虑DIC的可能;前列腺术后出血可有纤溶亢进因素参与。④颅内出血:通常见于血小板减少症患者,因中枢神经系统内丰富微血管之完整性需依靠血小板来完成;也见于凝血机制紊乱,但更多发生于复合因素所致的止凝血障碍。⑤阴道出血和月经过多:可为血小板减少首发表现,亦可见于纤溶亢进、抗凝物质增多者。
【临床诊断】
(一)毛细血管-血小板型止血缺陷
以皮肤、黏膜出血为主;有眼底和颅内出血,而肌肉、关节、内脏出血少见;创伤后,伤口即刻发生渗血难止,持续时间一般不长;压迫止血有效,止血后不易复发;遗传性最可能为血管性血友病;获得性最多见是ITP,其次是脾功能亢进。
(二)凝血障碍-抗凝物质型止血缺陷
以肌肉、关节和内脏出血为主,可伴皮肤黏膜出血,大面积瘀斑下常可触及血肿;创伤当时出血可能不明显,但延迟出血严重;出血持续时间较长,局部压迫和药物止血效果差,但输血或血制品有显效;遗传性常见为血友病(A、B);获得性多见于肝病,其次为维生素K缺乏、DIC、凝血因子抑制物、抗凝治疗。出血性疾病临床特征见表16-7-3。
(三)纤维蛋白溶解(纤溶)活性增强
以皮肤瘀斑,可融合成大片状地图样为特征;注射部位或创面渗血难止;凝血块易溶解;多为获得性或继发性。
表16-7-3 出血性疾病的临床特征

【实验诊断】
出血性疾病血液学检验的项目和手段已达到基因诊断水平。从众多检验项目中提出一组适用于初诊患者的过筛,并有助于进一步确定检验项目的选择,是十分必要的。
(一)毛细血管-血小板型止血缺陷
1.筛选试验
常用出血时间(BT国际标准化出血时间测定器法)和血小板计数(PLT)。
(1)BT和PLT均正常:
除正常人外,多数是血管性紫癜。
(2)BT延长伴PLT减少:
多数是血小板减少性紫癜。
(3)BT延长伴PLT增多:
多数是血小板增多症伴功能异常。
(4)BT延长伴PLT正常:
多数血小板功能异常或某些凝血因子缺陷所引致。
2.血小板功能检测
(1)黏附功能:
采用血小板黏附试验(PAdT)。
(2)聚集功能:
常用血小板聚集试验(PAgT)。
(3)释放反应:
β-TG、PF 4 、GMP-140、TSP、5-HT测定。
(4)花生四烯酸代谢:
TXB 2 、cAMP/cGMP测定等。
(5)收缩蛋白试验:
常用血块收缩时间。
(6)促凝活性:
PF 3 有效性(PF 3 aT)测定。
(二)凝血障碍-抗凝物质型止血缺陷
1.筛选试验
常用活化的部分凝血活酶时间(APTT)、血浆凝血酶原时间(PT)和凝血酶时间(TT)。APTT模拟内源性凝血过程,主要反映因子Ⅴ、Ⅷ、Ⅸ、Ⅹ、Ⅺ的变化;PT模拟外源性凝血过程,主要反映因子Ⅴ、Ⅶ、Ⅹ的变化;TT延长表明纤维蛋白原减少或血浆存在抗凝物质。
(1)APTT和PT均正常:
除正常人外,仅见于因子
 缺乏症。
缺乏症。
(2)APTT延长伴PT正常:
多数是内源凝血途径缺陷,若临床上有出血倾向,为血友病(A、B)、遗传性凝血因子Ⅺ缺乏症,循环中出现抗因子Ⅷ、Ⅸ或Ⅺ抗体,血管性血友病等;若临床上无出血倾向,则为因子Ⅻ、激肽释放酶原、高分子量激肽原缺乏。
(3)APTT正常伴PT延长:
多数是外源凝血途径缺陷,见于遗传性或获得性因子Ⅶ缺陷症,其中获得性者常见于肝病、维生素K缺乏、循环中因子Ⅶ抗体出现和口服抗凝剂等。
(4)APTT和PT均延长:
多数是由于共同途径凝血缺陷所引起的出血性疾病。如遗传性和获得性因子Ⅹ、Ⅴ、凝血酶原和纤维蛋白原缺陷症。获得性者主要见于肝病和DIC、口服抗凝剂、循环中抗凝血因子(Ⅹ、Ⅴ和凝血酶原)抗体出现及肝素治疗时。为明确病因,应进一步选择TT和纤维蛋白原(Fg)定量。①TT正常:Ⅹ、Ⅴ、凝血酶原缺陷,见于肝病、维生素K缺乏。②TT延长和Fg下降(<0.75g/L):低或缺乏纤维蛋白原血症。③TT延长和Fg正常(或不低于0.75g/L):血浆中存在抗凝物质。此时如果延长的TT可被甲苯胺蓝所纠正,表明肝素样物质增多;反之,表明FDP增多。
(5)凝血障碍与血小板异常共存:
如血管性血友病、DIC等。
对系统性止血障碍性疾病的初步筛查应该包括BT、PLT、APTT和PT。所有四项结果均正常,实际上已可排除任何有临床意义的系统性凝血功能障碍性疾病,但也有一些必须关注的例外。如因子
 缺乏症可有严重的出血倾向,但筛查试验是正常的;由于敏感性的原因,PT和APTT只能检出严重的、涉及因子水平在正常的30%以下的凝血因子缺陷。疑为轻度的凝血因子缺乏时,尚需做凝血因子水平(活性)检测。
缺乏症可有严重的出血倾向,但筛查试验是正常的;由于敏感性的原因,PT和APTT只能检出严重的、涉及因子水平在正常的30%以下的凝血因子缺陷。疑为轻度的凝血因子缺乏时,尚需做凝血因子水平(活性)检测。
PT和APTT延长除标明一种或以上的凝血因子缺乏外,尚有抑制物存在的可能,此种抑制物常为相应凝血因子抗体。区分这两种可能性,可行混合正常血浆纠正试验。
2.凝血因子检测
(1)凝血因子促凝活性(F∶C)和抗原性(F∶Ag)测定:包括Ⅱ、Ⅴ、Ⅶ、Ⅷ、Ⅸ、Ⅹ。
(2)vWF相关试验:vWF抗原测定(vWF∶Ag)、瑞斯托霉素辅因子活性测定(v WF∶RcoF)、瑞斯托霉素诱导的血小板凝集试验(RIPA)、vWF多聚体分析等。
(三)纤维蛋白溶解亢进出血的实验诊断
1.筛选试验
常用纤维蛋白(原)降解产物(FDP)和D-二聚体(D-D)测定。FDP是血液循环中纤维蛋白(原)在纤溶酶作用下生成的X(x)、Y(y)、D(d)、E(e)碎片,含量增高反映纤溶系统的激活。D-二聚体是交联纤维蛋白的降解产物,其表明纤维蛋白的形成及溶解的发生,理论上可用于原发性纤溶和继发性纤溶亢进的鉴别。
(1)FDP和D-D均正常:
表示纤溶活性正常。
(2)FDP阳性伴D-D阴性:
理论上只见于纤维蛋白原被降解,而未有纤维蛋白降解,见于原发性纤溶。实际上在肝病、术后大出血、重症DIC、纤溶初期、剧烈运动后、类风湿因子阳性、抗Rh(D)抗体存在条件下,可出现FDP假阳性。
(3)FDP阴性伴D-D阳性:
理论上只见于纤维蛋白被降解,见于血栓栓子自发性溶解。而在DIC、动静脉血栓形成和溶栓治疗后,可出现FDP假阴性。
(4)FDP和D-D均阳性:
纤维蛋白原和纤维蛋白同时被降解,见于继发性纤溶,如DIC和溶栓治疗。
2.纤溶功能检测
(1)t-PA(活性及抗原性)和u-PA测定。
(2)纤溶酶原活性(PLG:a)和抗原含量(PLG:Ag)测定。
(3)纤溶酶原激活抑制物(PAI)测定和α 2 -抗纤溶酶(α 2 -PI)测定。
(四)分子标志物检测
1.血管内皮细胞受损
血浆ET-1;TM抗原及活性检测。
2.血小板激活
β-TG;PF 4 ;GMP-140;TXB 2 。
3.凝血因子活化
F 1+2 ;FPA;可溶性纤维蛋白单体复合物(SFMC)。
4.抗凝和纤溶
凝血酶-抗凝血酶Ⅲ复合物(TAT);纤维蛋白肽Bβ 1-42 和Bβ 15-42 。前者为纤维蛋白原降解产物,后者见于纤维蛋白降解。
【治疗】
(一)毛细血管-血小板型止血缺陷
局部治疗包括压迫冷敷、凝血酶及吸收性明胶海绵应用;降低血管壁脆性和通透性的药物主要有:芦丁(rutoside),属黄酮类,可增强毛细血管壁抗力;卡巴克络(carbazochrome),可增强毛细血管及周围组织中的酸性黏多糖,降低血管壁通透性;酚磺乙胺(dicynone),增强血小板黏附、降低血管壁通透性;维生素C,作为羟化酶辅酶参与胶原组织中脯氨酸和赖氨酸羟化;肾上腺皮质激素可降低血管壁脆性和通透性。另外可选用血管收缩药如垂体后叶素等。
促进血小板生成药物如:血小板生成素(thrombopoietin TPO)可刺激骨髓巨核细胞生成发育成熟及血小板生成;白细胞介素11(IL-11)可促进巨核细胞成熟,增加外周血小板的数量;酚磺乙胺尚有促进血小板由骨髓释放作用。
增强血小板功能药包括:巴曲酶(血凝酶,batroxobin,reptilase)为血液凝固酶,促进血小板活化,诱导血小板聚集。
肾上腺皮质激素、免疫抑制剂、大剂量免疫球蛋白、脾切除对免疫性血小板减少症有效(见本篇第七章第五节)。
血小板输注,适应证为严重血小板减少症(≤20×10 9 /L)和(或)血小板功能缺陷。
(二)凝血障碍-抗凝物质型止血缺陷
维生素K参与因子Ⅱ、Ⅶ、Ⅸ、Ⅹ的合成。血浆及凝血因子制品主要有:新鲜冷冻血浆,指新鲜全血去红细胞于6小时内冷冻至-18℃,富含因子Ⅱ、Ⅴ、Ⅶ、Ⅸ、Ⅹ、Ⅺ、Ⅻ;冷沉淀物,新鲜冷冻血浆于4℃融化时产生,富含因子Ⅰ、Ⅷ、Ⅻ、vWF、Fn;纤维蛋白原制剂;因子Ⅷ浓缩物;vWF浓缩物;因子Ⅸ浓缩物;凝血酶原复合物浓缩物(PCC)富含因子Ⅱ、Ⅶ、Ⅸ、Ⅹ;其适应证包括严重肝病、血友病、维生素K缺乏、DIC,首次剂量一般为40U/kg,维持量为15~20U/(kg·d);凝血因子替代治疗并发症:过敏反应、凝血因子抗体、溶血性贫血、血栓形成、病毒性肝炎、AIDS等。凝血因子缺乏的补充治疗见表16-7-4。
表16-7-4 凝血因子缺乏的补充治疗
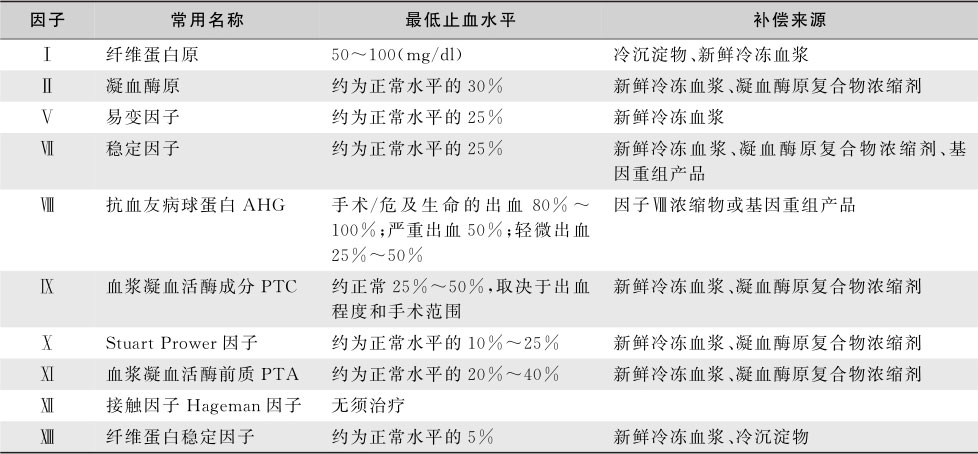
基因重组的FⅧ、FⅦ业已临床应用(见本章血友病节)。
针对病理性循环抗凝物质的治疗包括硫酸鱼精蛋白,适用于肝素过量、重症肝病;肾上腺皮质激素和免疫抑制剂的应用;补充大剂量凝血因子以中和抗体;血浆置换。
(三)纤维蛋白溶解活性增强
氨基己酸(EACA),竞争性抑制纤溶酶原与纤维蛋白的结合,使纤溶酶原不被其活化剂激活,其适应证有全身纤溶亢进(高纤溶酶血症、肝病、肿瘤、手术)、局部纤溶亢进(节育环月经过多、蛛网膜下腔出血、前列腺术后);同类药物有氨甲苯酸(PAMBA)和氨甲环酸(AMCA)。
其他具有综合作用的止血药物如1-去氨基-8-D-精氨酸加压素(DDAVP)、结合雌激素片(结合型雌激素)、促红细胞生成素。
主要参考文献
1.王振义,李家增,阮长耿.血栓与止血基础理论与临床.第3版.上海:上海科学技术出版社,2004.
2.王学锋,王鸿利.血栓与止血的检测及应用.上海:世界图书出版公司,2002.
3.Goldman L,Schafer AI.Goldman's Cecil Medicine.25th ed.Philadelphia:Elsevier Saunders,2016:1154-1158.
第二节
血管性紫癜
程韵枫 徐建民
血管性紫癜是指血管壁及周围组织异常所致的出血性疾病。血细胞从毛细血管内向外流出进入皮肤或皮下组织引起的损害统称为紫癜(purpura)。从血管内流出血液的量决定了皮肤损害的大小及范围。少量的出血产生针尖样大小的(<2mm)红色皮损称为瘀点,较多的出血(>10mm)引起的紫癜性损伤称为瘀斑。出血性损害的颜色取决于出血量的多少及部位,以及出血后经历的时间。浅表部位的出血初起通常为鲜红或深红色,较深部位的出血常常呈紫色,随着时间的推移可表现为深紫色、棕色、橘黄色或黄绿色。紫癜可由血小板数量减少、凝血因子缺乏、血小板功能异常及血管异常等因素引起。
血液从血管内流出进入皮肤或皮下组织的机制包括:①血管透壁压增加:如剧烈咳嗽、呕吐、分娩时用力、静脉淤滞等;②血管损伤:如紫外线辐射、感染、栓塞、过敏、炎症、肿瘤、中毒、药物相关性等因素对血管的损害;③微循环和血管支持组织的完整性降低:如年龄相关性紫癜、糖皮质激素使用过量、维生素C缺乏、结缔组织异常、淀粉样物质浸润等。
过敏性紫癜及遗传性出血性毛细血管扩张症将分别在本章第三节及第四节详述。
一、异常蛋白血症性紫癜
紫癜可发生于异常蛋白血症,但出血原因常是多方面的,包括血小板减少、血小板质量异常和凝血障碍、血黏度过高、异常蛋白直接损伤血管内皮细胞等。
(一)冷球蛋白血症
血清中存在冷球蛋白而引起的皮肤紫癜,四肢和其他身体受寒即可产生,也可发生于阴囊及臀部等处。有时呈荨麻疹样皮疹,中间为瘀点,伴瘙痒。慢性紫癜可有色素沉着,紫癜出现时常伴雷诺现象。
(二)巨球蛋白血症
由于巨球蛋白过多,血清黏稠度增高,出现皮肤紫癜,碰撞后易出血。也可发生视网膜出血、鼻出血以及黏膜出血,亦常可伴发冷球蛋白血症而发生雷诺现象,血清免疫电泳IgM明显增高是本病的特征。
(三)高丙种球蛋白血症性紫癜
是一种以下肢反复出现紫癜、血沉增速和血清丙种球蛋白多克隆IgG浓度增高为特点的异常球蛋白血症。多见于女性,起病隐袭,紫癜开始出现在两小腿及足部,可扩展至大腿、臀部及下腹部,特别在用力或机械损害的部位。紫癜可相互融合成瘀斑,可留有色素沉着。部分患者继发于其他全身性疾病,如干燥综合征、系统性红斑狼疮等。
(四)多发性骨髓瘤
异常浆细胞恶性增生,产生大量异常免疫球蛋白,在临床上可伴有一定的出血症状,出血原因主要是血小板生成减少、血小板功能障碍及凝血因子减少。另外,异常球蛋白浸润和损害血管壁,影响止血功能,引起血管性紫癜。
(五)淀粉样变性
由于淀粉样蛋白在组织和器官中沉着而引起相应病变的一种疾病,全身任何器官均可受累,淀粉样蛋白一般先在毛细血管及小动脉周围开始沉着,使毛细血管脆性增高。紫癜常出现在面部和颈部。本病可以为原发性,也可以继发于其他疾病。
治疗主要针对原发病。
二、单纯性紫癜
也称为女性易青紫综合征(female easy bruising syndrome)。发病以青年女性为主,常与月经周期有关,可能与激素对血管和周围组织的影响有关,若同时服用影响血小板功能的药物如非甾体类抗炎药可使此类紫癜加重。主要表现为轻微创伤后或自发性出现下肢皮肤紫癜或瘀斑,病变局限于皮肤,常反复发作,但不留后遗症。束臂试验检查往往阳性。避免使用非甾体类抗炎药,必要时可服维生素C、芦丁和卡巴克络(安络血)等。临床上注意与轻度血小板病、血管性血友病或血小板功能异常相鉴别。
三、药物性紫癜
大多数与药物引起血小板减少有关,但少数情况下药物可引起紫癜而血小板数量正常。可能的机制是免疫复合物引起小血管损伤、削弱血管周围的支撑组织和干扰血小板的功能。部分患者在注射香豆素及其衍生物后会出现出血性皮肤梗死,提示暴发性紫癜可能与蛋白C和蛋白S缺乏有关。阿司匹林、吲哚美辛、别嘌醇、磺胺药、青霉素、奎宁及香豆类等药物均可引起皮肤紫癜。治疗应停用相关药物,或给予维生素C及糖皮质激素等。
四、感染性紫癜
细菌、病毒、立克次体和原虫感染均可引起紫癜,常见于流行性脑脊髓膜炎、败血症、出血热、伤寒、斑疹伤寒及疟疾等。产生机制是免疫复合物或病原体直接损伤内皮细胞,或使毛细血管通透性增加,或是毛细血管细菌性栓塞,部分病例是由多因素引起。治疗以抗感染为主。儿童在感染后全身皮肤可见大片触痛性瘀斑,伴有出血性大疱和坏死,内脏亦可受累,死亡率较高,称为暴发性紫癜。
五、维生素C缺乏性紫癜
既往称为坏血病(scurvy)。由于胶原分子中的羟脯氨酸的合成需要维生素C,故维生素C缺乏影响胶原合成,使血管壁及周围的结缔组织的韧性降低,毛细血管脆性增加而引起出血。表现为毛囊周围皮肤出血,肌肉出血,牙龈肿胀和皮肤角化过度。治疗可口服维生素C 200mg/d。
六、自身红细胞致敏性紫癜
本病少见,仅见于中年女性,常有损伤史。特点是自发性触痛性瘀斑,发病机制不明。本病过去认为与神经精神因素有关,也有人称为“精神性紫癜”,发病前常有明显情绪诱发因素,具有歇斯底里或其他精神异常的患者有发生本病的倾向,但目前认为与红细胞致敏有关。先局部瘙痒、刺痛或烧灼感,随后瘀斑周围可有红斑和水肿,并逐渐扩大出现疼痛性红斑。同时伴有头痛、发热、恶心、呕吐和腹痛等。出血可从肢体扩张到其他部位,可有黑便、血尿甚至颅内出血。出血常可自行停止,但可复发。病程迁延数年。各种出凝血检查均属正常。皮下注射自身全血、洗涤红细胞、自身血红蛋白,甚至注射由自身红细胞分离出的磷脂酸-L-丝氨酸可引起类似发作时出现的瘀斑。对症处理和精神治疗有一定疗效。
七、自身DNA致敏性紫癜
多见于女性,既往无外伤史。临床经过同自身红细胞致敏性紫癜。病损先出现在四肢,表现为皮肤疼痛性硬结和瘀斑,随后迅速增大和变硬。数天或2~3周内痊愈。除束臂试验可阳性外,其他出凝血检查均属正常。皮下注射患者的白细胞DNA溶液可使紫癜发作。本病病程较为迁延可持续数年,可有反复和加重。氯喹治疗有效,疗效迅速而显著,但停药后易复发。剂量为0.25g,每天4次,连续服用1周,以后减量为每天0.25~0.50g。精神治疗有一定疗效,但很少获得永久性的缓解。
八、老年性紫癜
老年人由于皮下结缔组织中胶原、弹性硬蛋白、脂肪等组织萎缩、松弛,以致血管床周围依托不足,轻度外伤或皮肤移动牵拉小血管即可引起皮下出血及瘀斑。紫癜常见于面部、颈部、手背、前臂和小腿,亦可轻微外伤后出现紫癜,可持续数周,吸收缓慢,以后留下棕色色素沉着。束臂试验阳性,其余检查均正常。
九、遗传性血管性紫癜
如Marfan综合征、Ehlers-Danlos综合征及弹性假黄瘤等,由于结缔组织基质发育异常使血管脆性增加引起皮肤出血。尚无有效治疗方法。
十、其他血管性紫癜
还有机械性紫癜、人为紫癜、直立性紫癜、激素性紫癜以及医源性紫癜等。
第三节
过敏性紫癜
程韵枫 徐建民
过敏性紫癜(anaphylactoid purpura)又称出血性毛细血管中毒症或 Henoch-Schönlein紫癜(HSP),是一种较常见的毛细血管变态反应性出血性疾病。临床以血液逸于皮肤、黏膜之下,出现瘀点瘀斑为常见,可累及皮肤、胃肠道、肾脏、关节,甚至心、脑等多个器官,出现肿痛、腹痛、便血、血尿和蛋白尿。过敏性紫癜的病因及发病机制仍未完全阐明,可能涉及感染、遗传、药物、疫苗及某些食物诱发等因素,以体液免疫异常为主,T淋巴细胞功能改变以及细胞因子等在发病中起重要作用。本病多见于儿童和青少年。平均年龄为5岁,男女之比为3∶2。
【病因与发病机制】
由于机体对某些过敏物质发生变态反应而引起毛细血管壁的通透性和脆性增高。与本病发生有关的因素有:①感染:细菌以溶血性链球菌多见,其他细菌如金黄色葡萄球菌、结核分枝杆菌和肺炎球菌亦可引起;幽门螺杆菌Hp感染可能是HSP发病的重要因素之一,根治Hp有利于HSP的康复,特别是腹型HSP患者。柯萨奇病毒、EB病毒、微小病毒B19、麻疹病毒、风疹病毒、水痘病毒以及肝炎病毒等感染也可诱发HSP。寄生虫侵入机体后其代谢产物或死亡后的分解产物都是异性蛋白质,可引起本病。②药物:青霉素、链霉素、磺胺类、异烟肼、水杨酸钠、奎宁等。③食物:鱼、虾、蟹、蛋、牛奶、鸡以及海鲜等异性蛋白质。④疫苗接种:流感疫苗、乙肝疫苗、狂犬疫苗、流脑疫苗等的接种可能诱发HSP。⑤遗传因素:有关遗传学研究涉及的基因主要有 HLA,可能与DRB*01,DRB l*11,DRB1*14及HLA-B35等型别相关;家族性地中海基因、血管紧张素转换酶基因(ACE基因)、血管内皮生长因子基因、PAX2基因等均可能与HSP的发病相关。⑥其他:如寒冷、外伤、花粉吸入、更年期甚至精神因素都能诱发本病。
以上各种因素引起自身免疫反应,免疫复合物反应损害小血管,发生广泛的毛细血管炎,甚至坏死性小动脉炎,造成血管壁通渗性和脆性增高,导致皮下组织、黏膜及内脏器官出血及水肿。体液免疫异常:HSP发病多为IgA所介导,在HSP患者血清中往往可检测出IgA循环免疫复合物,且多沉积于表皮血管壁,有报道约50%的HSP患者血清IgA水平升高;HSP患者血清IgE水平升高,免疫复合物沉积或由于IgE介导血管损伤,导致毛细血管和小血管壁及其周围组织产生炎症,血管壁通透性增高,从而产生紫癜和各种局部或全身症状。电镜检查肾小球血管系膜有免疫复合物沉着,经免疫荧光证明主要是IgA(少量为IgG及IgM)、C3、纤维蛋白/纤维蛋白原,故过敏性紫癜肾脏损害与免疫复合物有关。细胞免疫异常:HSP患者B细胞处于高激活状态,T细胞功能紊乱。T细胞功能紊乱主要表现在T细胞亚群失调、活化异常及调节性T细胞功能紊乱。HSP急性期患者外周血CD4 + 数量降低,CD8 + 数量升高,CD4 + /CD8 + 比值下降,Th1/Th2失衡,主要向Th2亚群分化,CD4 + CD25 + 调节性T细胞显著降低。Th17在HSP患者中显著高于健康对照组,表明Th17的异常活化与HSP的发生及发展相关。细胞因子IL-6、IL-4、TNF-α在HSP患者血清中表达水平显著升高,可能是致HSP发病的重要原因;血管内皮生长因子(VEGF)明显增高,血清、尿液可溶性黏附分子-1(sICAM-1)和可溶性血管细胞黏附分子-1(sVCAM-1)水平在HSP急性期显著高于恢复期和健康对照,提示尿sICAM-1和sVCAM-1水平高低可能与HSP患者肾脏病变程度有关;补体水平下降。
【临床表现】
本病主要见于儿童及青年,6岁以上占多数。春秋季节好发。起病前1~3周有上呼吸道感染史。可有倦怠、乏力、低热、纳差等前驱症状。
(一)皮肤
首起症状以皮肤紫癜最常见。多在前驱症状2~3天后出现,常对称性分布,以下肢伸侧及臀部多见,分批出现,紫癜大小不等,呈紫红色,略高出皮肤,可互相融合,常伴荨麻疹、多形性红斑及局限性或弥漫性水肿,偶有痒感。严重的紫癜可融合成大疱,发生中心出血性坏死。皮肤损害有4种类型:①单纯性紫癜常伴天疱疮样皮肤损害。②荨麻疹伴血管神经性水肿。③弥散性红斑,伴或不伴水肿。④皮肤坏死,或伴溃疡形成。
(二)腹部
约50%的病例有腹痛,常发生在出疹的1~7天,位于脐周或下腹部,呈阵发性绞痛,可有压痛但无肌紧张,呈症状与体征分离现象。严重者可合并呕吐及消化道出血(呕血、便血等)。由于肠蠕动紊乱,可诱发肠套叠,在小儿多见。肠坏死、肠穿孔者少见。少数患者可误诊为急腹症而进行剖腹探查。
(三)关节症状
多见于膝、踝等大关节,呈游走性,可有轻微疼痛或明显的红、肿、痛及活动障碍,反复发作,但不遗留关节畸形,易误诊为风湿性关节炎。
(四)肾脏病变
见于1/2~1/3的患者,一般于紫癜出现后1~8周内发生,可持续数月或数年,主要表现为血尿、蛋白尿、水肿、高血压。个别严重病例死于尿毒症。
(五)神经症状
当病变累及脑和脑膜血管时,可出现各种神经系统症状,如头痛、头晕、呕吐、目眩、神志恍惚、烦躁、谵妄、癫痫、偏瘫、意识模糊、昏迷等,但例数极少。
(六)其他症状
病变累及呼吸道时,可出现咯血、胸膜炎症状,临床少见。
根据体征可将本病分为皮肤型(单纯紫癜型)、腹型(Schönlein型)、关节型(Henoch型),若有两种以上并存时称为混合型。
【实验室检查】
(一)一般检查
白细胞计数正常或轻度升高,有感染时可增高。合并寄生虫感染者嗜酸性粒细胞可增高。红细胞和血红蛋白一般正常或轻度降低。合并内脏出血者可呈中度失血性贫血,血小板计数多数正常。尿常规结果取决于肾脏受累程度,若伴发肾炎时,血尿和蛋白尿极为常见,偶尔可见管型尿。大便可找到寄生虫或虫卵,胃肠受累时大便隐血阳性。血沉增高见于2/3的病例,血清循环免疫复合物(CIC)增高。在严重肾型病例,尿素氮及肌酐增高。患者的骨髓象检查均正常。划痕试验可以阳性。
(二)出凝血机制检查
30%~50%的病例束臂试验阳性。甲皱毛细血管检查可见到毛细血管扩张、扭曲或畸形,对针刺反应也减弱。其他检查如出血、凝血时间和血块收缩等均在正常范围。
(三)免疫学检查
约50%的病例的血清IgG和IgA增高,但以IgA增高为明显。有些病例IgE增高,临床无特异性。
(四)皮肤或肾脏活检
行病理组织学或电子显微镜检查对非典型病变具有重要诊断价值。
【诊断与鉴别诊断】
(一)诊断
典型病例的诊断并不困难,凡是有下列特点者即可诊断:①四肢出现对称分布、分批出现的紫癜,特别以下肢为主。②在紫癜出现前后,可伴有腹部绞痛、便血、关节酸痛、血尿及水肿等。③血小板计数、凝血功能检查及骨髓检查等均正常。
(二)鉴别诊断
1.皮肤型需与药疹或血小板减少性紫癜作鉴别。药疹患者有服药史,皮疹常分布于全身,停药后药疹即可消失。血小板减少性紫癜患者瘀点和瘀斑可呈不规则分布,皮疹不隆起,无丘疹荨麻疹,血小板计数减少,骨髓象见巨核细胞成熟障碍。
2.关节型需与风湿病鉴别。
3.腹型本病腹痛部位不固定,腹痛虽明显,但局部体征较轻,且多无腹肌紧张。需与急性阑尾炎、肠梗阻、肠套叠、肠穿孔等鉴别。
4.肾型需与急性肾小球肾炎、狼疮肾炎、肾结核等区别。肾小球肾炎患者无皮肤紫癜及腹部、关节症状。狼疮肾炎患者有多脏器损害、白细胞减少、血沉增快、狼疮细胞阳性及其他免疫指标阳性。
【治疗】
(一)消除致病因素
原则上应停止接触任何可能引起过敏的物质,停用可能引起过敏的食物或药物,去除病灶,控制感染,驱除寄生虫。
(二)一般治疗
1.抗组胺类药物
可选用盐酸异丙嗪(非那根)每次12.5~25.0mg,每天3次,口服:马来酸氯苯那敏(扑尔敏)每次4mg,每天3次,口服;苯噻啶每次0.5~1.0mg,每天1~3次,口服;盐酸去氯羟嗪(克敏嗪)每次25~50mg,每天3次,口服;特非那定每次60mg,每天2次,口服;氯雷他定(克敏能),每次10mg,每天l次,口服;阿司咪唑(息斯敏),每次10mg,每天1次,口服。10%葡萄糖酸钙静脉注射也可应用,但疗效不肯定。
2.芦丁和维生素C
作为辅助用药,芦丁10~15片,每天3次,口服;维生素 C1g,每天 1~2次口服或静脉滴注。
3.止血药
卡巴克洛(安络血)每次10mg,每天2~3次,肌内注射,或用40~60mg加入葡萄糖溶液中静脉滴注;酚磺乙胺(止血敏)0.25~0.5g,每天2~3次肌内注射、静脉注射或静脉滴注。有肾脏病变者,抗纤溶药物应慎用。
(三)肾上腺皮质激素
对关节型、腹型和皮肤型疗效较好,对肾型无效,不能改变肾型患者预后。常用泼尼松每天30mg,直至紫癜消失后逐渐停药。如1周后皮疹不退可加至每天40~60mg。病情急重者可用氢化可的松每天100~200mg,静脉滴注,待病情好转后改用口服。
(四)免疫抑制剂
如以上疗法效果不佳时可试用免疫抑制剂,特别是合并肾脏损害的病例。环磷酰胺2.5mg/(kg·d),口服,或硫唑嘌呤2.5mg/(kg·d),口服,连续4~6个月。免疫抑制剂也可与肾上腺皮质激素合用。
(五)抗凝治疗
急进性肾炎、肾病综合征病例,除用皮质激素、环磷酰胺外,还可用抗凝治疗。如肝素治疗使APTT维持至正常值的1.5~2.0倍;后改华法林10~20mg/d,以3~5mg/d维持,使凝血酶原时间维持在正常的1~2倍。
【病程与预后】
病程一般为3~6个月,一次发作可持续1周~6个月不等。关节及皮肤症状者病期较短,腹部症状明显者病程较长,病原持续存在时可转为慢性。如再次接触过敏原,可导致本病复发。本病多呈自限性,一般6周内可自愈,预后大多良好,少数迁延数年。如果并发肾炎进展为肾衰竭,或有脑部病变并发脑出血者,预后不良。
第四节
遗传性出血性毛细血管扩张症
程韵枫 徐建民
遗传性出血性毛细血管扩张症(hereditary hemorrhagic telangiectasia,HHT)是一种常染色体显性遗传性疾病,其特征为皮肤、黏膜多部位的毛细血管扩张性损害,引起鼻出血和其他部位出血。日本的发病率约为1/8000~1/5000。国内尚缺乏临床流行病学的资料统计。
【病因与发病机制】
本病系常染色体显性遗传。可能与HHT发病相关的两个基因,分别是位于染色体9q33-34的 endoglin 与位于12q的 ALK1 ,这两个基因分别表达转化生长因子-β(TGF-β)的Ⅲ型和Ⅰ型受体。但是目前这两个基因缺陷与本病的发病关系尚未完全阐明。
本病的主要病理变化是血管缺乏弹性纤维和平滑肌,受累的血管壁仅仅只一层内皮细胞,其周围由一层无肌肉、无弹性的疏松结缔组织所包围。血管壁脆弱易破,已不能收缩,呈血管瘤样扩张,称为毛细血管扩张,故容易引起出血。
【临床特点】
男女皆可发病。病变以皮肤与黏膜血管的表层处最常见,多见于甲床和手部皮肤、颜面、阴囊、鼻、嘴唇、舌部、消化道等。病变处可见成簇的红色或紫色斑点,或呈圆形,散在的或孤立的小血管瘤,稍微隆起于皮肤,直径为1~2mm,分界明显,用玻片加压即褪色,但褪色不完全。在肺部出现动静脉畸形(PAVM),其他部位也可出现动静脉瘘。本病的主要临床表现为同一部位的反复出血,出血症状常在幼年即出现,但也有在中年或老年以后才首次出现者。常随年龄增长加重。在40~60岁之间可达高峰。
【实验室检查】
血小板计数、各种出血及凝血试验无明显异常,但束臂试验可阳性。甲皱毛细血管镜检查可发现高度扩张与扭曲成团的血管袢,且对针刺无收缩反应。合并PAVM者,胸部X线检查可能发现一种“钱币”样阴影,但微小的病变常常被遗漏。螺旋CT扫描诊断PAVM的敏感性较高。对消化道出血、血尿、咯血等内脏出血的患者,在做相应的内镜检查时,在黏膜表面可见到扩张的毛细血管。
【诊断】
诊断主要根据:①反复发作的自发性鼻出血;②多个特征部位出现毛细血管扩张,如唇、鼻、手指和口腔黏膜等;③内脏受累。如消化道的毛细血管扩张,肺、肝、脑的动静脉畸形;④阳性家族史,直系亲属中有HHT患者。符合以上3条或3条以上条件者可确诊为HHT,符合其中2条者为疑似病例。少于2条者暂不考虑HHT。
【治疗】
本病无特殊治疗方法,以对症治疗为主。鼻出血可用吸收性鼻腔填塞物,或加压止血处理,可同时加用止血剂。严重反复的出血可采用激光、冷冻或手术缝合等措施,但易复发。对于PAVM,可采用肺叶切除或栓塞疗法。女性绝经期后常有出血加重的趋向,有用雌激素治疗鼻出血的报道。避免外伤,避免服用阿司匹林类药物,也应避免引起血压增高及血管扩张的因素和药物。
主要参考文献
1.邓家栋,杨崇礼,杨天楹,等.邓家栋临床血液学.上海:上海科学技术出版社,2001.
2.Yang YH,Yu HH,Chiang BL.The diagnosis and classification of Henoch-Schönlein purpura:an updated review.Autoimmun Rev,2014,13(4-5):355-358.
3.Audemard-Verger A,Pillebout E,Guillevin L,et al.IgA vasculitis(Henoch-Shönlein purpura)in adults:Diagnostic and therapeutic aspects.Autoimmun Rev,2015,14(7):579-585.
第五节
原发免疫性血小板减少症
程韵枫
原发免疫性血小板减少症(primary immune thrombocytopenia,ITP)是一种获得性免疫介导的血小板减少疾病。目前定义为外周血小板少于100×10 9 /L,没有其他引起血小板减少的诱因或基础疾病。ITP是较为常见的出血性疾病,年发病率(5~10)/10 6 ,育龄期女性发病率高于男性,其他年龄阶段,ITP发病率男女比例无差别。在成年人,典型病例一般为隐匿发病,病前无明显的病毒感染或其他疾病史,病程为慢性过程。在儿童,一般病程短暂,80%的患儿在6个月内自发缓解。
【病因与发病机制】
ITP的经典发病机制是自身抗体致敏的血小板被单核-巨噬细胞系统过度破坏所致,近年来细胞免疫介导的血小板减少亦在ITP的发病中起着重要作用。该病的主要发病机制:患者对自身抗原的免疫失耐受,导致免疫介导的血小板破坏增多和免疫介导的巨核细胞产生血小板的相对不足。
50%~60%的ITP患者血小板表面包被有IgG型自身抗体,可识别血小板表面的一种或多种糖蛋白(GP),包括GPⅡb/Ⅲa、GPⅠb/Ⅸ、GPⅠa/Ⅱa、GPⅥ等,其中约75%的血小板抗原都位于血小板膜GPⅡb/Ⅲa或GPⅠb/Ⅸ复合体上。儿童ITP的发病可能与病毒感染密切相关,其中包括疱疹病毒、EB病毒、巨细胞病毒、微小病毒B19、麻疹病毒、流行性腮腺炎病毒、风疹病毒和肝炎病毒等。通常在感染后2~21天发病。ITP患者自身抗体的产生机制包括了血小板、抗原递呈细胞(antigen-presenting cells,APCs)、T细胞和B细胞之间的相互作用。①APCs捕获血小板抗原,加工处理成抗原肽并提呈到APCs表面;②活化的APCs把抗原肽提呈给CD4 HLA-DR限制性T细胞 和CD3/CD8 T细胞;③CD4HLA-DR限制性T细胞活化,产生细胞因子,刺激B细胞分化产生抗体;④APCs和CD4HLA-DR限制性T细胞,以及CD4HLA-DR限制性T细胞与B细胞之间的相互作用被CD40、CD154等共刺激因子加强并导致特异性T细胞和B细胞克隆性增生,从而导致最初的免疫反应维持与放大;⑤活动期ITP患者,活化的T淋巴细胞对凋亡的抵抗,从而产生持续的免疫性血小板破坏;⑥克隆性增生的B细胞产生血小板特异性抗体,结合了抗体的血小板一方面与脾巨噬细胞表面的Fcγ受体结合,血小板被吞噬;⑦血小板本身也表达CD154(CD40L)与APCs表面的CD40相互作用,进一步导致自身免疫反应放大。
细胞免疫异常在ITP发病中也起着非常重要的作用,ITP患者CD4 + T细胞亚群异常,主要表现为Th1/Th2的比率失衡,主要向Th1亚群分化,能抑制自身反应性T、B细胞的活化和增殖的CD4 + CD25 + 调节性T细胞减少。细胞毒性T细胞(cytotoxic T cell,CTL)介导的针对血小板的细胞毒作用是ITP患者血小板破坏的原因之一,FasL、TNF-α与相应受体结合所介导的凋亡途径是CTL发挥其细胞毒作用的机制之一,另外穿孔素和颗粒酶B途径参与了CTL对血小板的细胞毒作用。
传统的ITP发病机制认为,ITP患者表现为骨髓巨核细胞数升高,然而,对ITP患者骨髓形态学的研究发现,多数患者骨髓巨核细胞计数并不增多,形态异常,如胞质空泡增多、颗粒减少、细胞膜光滑等,且巨核细胞超微结构异常,包括由于线粒体和内质网肿胀形成的胞质内空泡增多以及染色质固缩等,显示广泛凋亡。抗血小板抗体可与巨核细胞发生特异性结合,可干扰巨核细胞的成熟、血小板产生及释放。T细胞亦可能介导巨核细胞的增生及凋亡异常。
【临床表现】
儿童新诊断的ITP发病前1~3周84%的患者有急性上呼吸道或其他病毒感染史,部分发生在预防接种之后,起病急,血小板显著减少,可有轻度发热、畏寒,突然发生广泛而严重的皮肤黏膜紫癜,甚至大片瘀斑。黏膜出血多见于鼻腔、齿龈,口腔可有血疱。胃肠道及泌尿道出血并不少见,不到1%的患儿发生颅内出血而危及生命。如患者头痛、呕吐,则要警惕颅内出血的可能。80%以上的患者可自行缓解,少数可迁延转为慢性。
ITP患者的出血表现在一定程度上与血小板计数有关,血小板数在(20~50)×10 9 /L之间轻度外伤即可引起出血,少数为自发性出血,如瘀斑、瘀点等,血小板数<20×10 9 /L有严重出血的危险。皮肤紫癜、瘀斑、瘀点多见,静脉穿刺点周围可见瘀斑,一般无皮下或关节血肿。可有鼻、牙龈及口腔黏膜出血,口腔血疱见于严重血小板减少,女性月经过多有时是唯一症状,泌尿道及胃肠道出血分别表现为血尿及黑粪,呕血少见。脾通常不大。
【实验室检查】
(一)外周血细胞计数
ITP的特点是外周血只有血小板减少而其他各系血细胞都在正常范围。部分患者由于失血导致缺铁,可有贫血存在。单纯ITP网织红细胞计数基本正常。
(二)外周血涂片
由于EDTA依赖性血小板凝集而导致的假性血小板减少需排除;出现破碎红细胞应除外血栓性血小板减少性紫癜(thrombotic thrombocytopenic purpura,TTP)和溶血尿毒综合征(hemolytic uremic syndrome,HUS);过多出现的巨血小板或微小血小板需考虑遗传性血小板减少症。
(三)骨髓检查
ITP骨髓呈增生象,巨核细胞数可正常或增多,有成熟障碍,产血小板的巨核细胞数明显减少。
(四)HIV和丙型肝炎病毒(HCV)检测
对疑似ITP的成人患者均应进行HIV和HCV检查,HIV及HCV感染引起的血小板减少在临床上有时很难与原发性ITP患者相鉴别。
(五)免疫球蛋白定量
血清IgG、IgA、IgM水平要求常规测定。低水平的免疫球蛋白常提示常见变异型免疫缺陷病(CVID)或选择性IgA缺陷症。
(六)其他实验室检查
抗GPⅡb/Ⅲa、GPⅠb/Ⅸ等抗血小板自身抗体的测定有助于鉴别免疫性和非免疫性血小板减少,目前采用MAPIA法检测。贫血伴有网织红细胞计数升高时应做直接抗人球蛋白试验(DAT)。ITP患者通常血清血小板生成素(TPO)水平正常,网织血小板计数增加。儿童ITP患者抗核抗体(ANA)阳性可能预示为慢性。测定甲状腺功能、甲状腺球蛋白抗体和促甲状腺激素(TSH)对鉴别患者是否出现临床甲状腺疾病有意义。部分慢性感染,如微小病毒和巨细胞病毒(CMV)也出现血小板减少。血清学或 14 C呼气试验可确定是否存在幽门螺杆菌(HP)感染。
【诊断与鉴别诊断】
(一)目前ITP的诊断仍是临床排除性诊断
根据病史,家族史,皮肤、黏膜出血症状,其诊断要点如下:①至少2次检查血小板计数减少,血细胞形态无异常;②脾一般不大;③骨髓中巨核细胞数正常或增多,伴有成熟障碍;④须排除其他继发性血小板减少症。
由药物引起的血小板减少应仔细询问服药史;先天性血小板减少性紫癜与本病相似,应调查家族史,必要时检查其他家庭成员加以区别;结缔组织疾病早期的表现可能仅有血小板减少,对血小板减少患者应进行相关实验室检查;伴有血栓形成者注意抗磷脂综合征,应询问流产史及检测抗磷脂抗体加以鉴别;伴有溶血性贫血者应考虑为Evans综合征;伴有中度以上脾大者应考虑脾功能亢进,除血小板减少外还有白细胞减少及贫血;血涂片中出现红细胞碎片提示血小板减少可能与血栓性微血管病有关;DIC患者有多项凝血功能检查异常;获得性单纯无/低巨核细胞性血小板减少症患者可仅表现为血小板减少,但其骨髓中巨核细胞阙如或减少;白血病、淋巴系统增殖性疾病、骨髓瘤及骨髓增生异常综合征(MDS)等均可有血小板减少,骨髓检查可资鉴别;此外HIV感染者亦有血小板减少,可通过检测HIV抗体及CD4 + 细胞数值下降等加以鉴别;对HP感染阳性者予以抗HP治疗,若治疗有效者血小板计数上升则为HP所致的继发性ITP;TPO有助于鉴别ITP与不典型再生障碍性贫血(AA)或低增生性MDS。
(二)按疾病发生的时间及其治疗情况分期
1.新诊断的ITP
指确诊后3个月以内的ITP患者。
2.持续性ITP
指确诊后3~12个月血小板持续减少的ITP患者。包括没有自发缓解的患者或停止治疗后不能维持完全缓解的患者。
3.慢性ITP
指血小板减少持续超过12个月的ITP患者。
4.重症ITP
指PLT<10×10 9 /L,且就诊时存在需要治疗的出血症状或常规治疗中发生新的出血症状,且需要采用其他升高血小板药物治疗或增加现有治疗的药物剂量。
5.难治性ITP
指满足以下3个条件的患者:①脾切除后无效或者复发;②仍需要治疗以降低出血的危险;③除外其他原因引起的血小板减少症,确诊为ITP。
【治疗】
ITP的治疗应个体化。一般说来血小板计数>50×10 9 /L、无出血倾向者可予观察并定期检查;血小板计数介于(20~50)×10 9 /L之间,则要视患者临床表现、出血程度及风险而定;血小板<20×10 9 /L者通常应予治疗。出血倾向严重的患者应卧床休息,避免外伤,避免服用影响血小板功能的药物。本病治疗的目的是控制出血症状,减少血小板的破坏,但不强调将血小板计数提高至正常,以确保患者不因出血发生危险,又不因过度治疗而引起严重不良反应。
(一)ITP的初始治疗
1.肾上腺糖皮质激素治疗
糖皮质激素是标准的初治治疗,但应注意其副作用。泼尼松,常用起始剂量为1mg/(kg·d),亦可选用泼尼松龙或氢化可的松。有反应的患者一周内血小板开始上升,2~4周内达峰值,稳定后逐渐减量至5~10mg/d维持,3~6个月后停药,维持用药最多不超过1年。约70%~90%的患者有不同程度的缓解,15%~50%的患者血小板恢复正常。治疗4周后血小板仍低于30×10 9 /L且增加不到基础值的两倍者,表明激素无效。短程地塞米松治疗方法为40mg/d,共4天,约85%的患者有效。若在6个月内血小板计数再次降至20×10 9 /L以下,则可重复一次,然后泼尼松15mg/d维持,并渐减量。
2.大剂量丙种球蛋白(IVIG)
IVIG通过封闭单核-巨噬细胞上的Fc受体,抑制抗体与血小板的结合。剂量为400mg/(kg·d),静脉滴注,连续5天,或1000mg/(kg·d),连用2天。24小时内即可见效,1周后血小板达到最高水平,有效率约75%,但疗效短暂,血小板计数在1个月内便降至原来水平。
(二)急诊治疗
对于出血风险高或需要急诊手术的血小板减少患者需要立即升高血小板。糖皮质激素加IVIG作为推荐疗法,大剂量甲泼尼龙(HDMP)(1g/d,×3天),可与IVIG联合应用。其他快速有效的治疗包括血小板输注。应用抗纤溶药物氨甲环酸1g,3次/日或氨基己酸1~4g,每4~6小时1次,最大剂量24g/d,可阻止严重血小板减少患者的反复出血。
(三)ITP的二线治疗
1.脾切除
是治疗本病有效的方法之一。作用机制是减少血小板抗体生成,消除血小板破坏的场所。切除指征:①经糖皮质激素和其他内科治疗无效,病程超过6个月以上者;②激素治疗虽然有效,但对激素产生依赖,停药或减量后复发,或需较大剂量(泼尼松30mg/d以上)维持才能控制出血者;③激素治疗有禁忌证;④有颅内出血倾向,经内科治疗无效者。手术相对禁忌证有:①ITP首次发作;②患有心脏病等严重疾病,不能耐受手术;③妊娠妇女患ITP;④儿童患者,尤其是5岁以下患儿切脾后可发生难以控制的感染。有条件者术前可用放射性核素示踪方法了解血小板破坏的主要场所是否在脾。切脾有效者术后出血迅速停止,术后24~48小时内血小板上升,10天左右达高峰,70%~90%的患者可获得明显疗效,其中约60%的患者获得持续完全缓解。腹腔镜脾切除具有创伤小、切口小、恢复快等优点。术前、术中认真检查有无副脾,切脾无效或复发时可再用激素治疗。
2.可供选择的二线治疗药物
硫唑嘌呤1~3mg/(kg·d);环孢素3~5mg/(kg·d),分两次口服,血药浓度控制在100~200ng/ml,用药期间监测肝肾功能;重组人血小板生成素(rhTPO)剂量1.0μg/(kg·d)×14天,血小板计数>100×10 9 /L时停药;达那唑口服10~15mg/(kg·d),反应率为60%~67%,老年女性和脾切除术后患者反应率最高;吗替麦考酚酯(吗替麦考酚酯,MMF)是一种抗增生性免疫抑制剂,MMF治疗需逐渐增加剂量(250mg逐渐增加到1000mg/d,2次/周,至少3~4周);利妥昔单抗375mg/m 2 ,每周1次,共4次,一般在注射后4~8周内起效;罗米司亭(romiplostim)和艾曲波帕(eltrombopag)作为TPO受体激动剂,其作用方式是刺激血小板生成,罗米司亭1~10μg/kg每周1次皮下注射。艾曲波帕是一种口服的非肽类TPO受体激动剂,每日给予25~75mg;TPO受体激动剂在脾切除术后患者中总有效率接近79%;长春新碱总剂量6mg,每周注射1~2mg,共4次。
(四)一线和二线治疗失败ITP患者的治疗
大约20%的患者在应用一线和二线治疗或行脾切除术后不能达到可以止血的血小板数,另外,10%~20%对脾切除术有效的患者最终复发。慢性难治性ITP可以选择环磷酰胺、联合化疗、吗替麦考酚酯及干细胞移植等治疗。
Campath-1H(阿仑珠单抗)可作为严重的难治性ITP选择性治疗的一种方式。该药有导致严重的有可能危及患者生命的免疫抑制的可能,因此需要长期的抗真菌、抗细菌和抗病毒的预防性治疗。自体或异体造血干细胞移植(HSCT)只被应用于严重的慢性难治性ITP且其他治疗方式无效的患者。
主要参考文献
1.中华医学会血液学分会血栓与止血学组.成人原发免疫性血小板减少症诊治的中国专家共识(2016版).中华血液学杂志,2016,37(02):89-93.
2.Cines DB,Bussel JB,Liebman HA,et al.The ITP syn-drome:pathogenic and clinical diversity.Blood,2009,113(26):6511-6521.
3.Rodeghiero F,Stasi R,Gernsheimer T,et al.Standardiza-tion of terminology,definitions and outcome criteria in immune thrombocytopenic purpura of adults and children:report from an in-ternational working group.Blood,2009,113(11):2386-2393.
第六节
继发性血小板减少症
化范例 程韵枫
继发性血小板减少症(secondary thrombocytopenia)是指有明确病因或在一些原发病基础上发生的血小板减少症。
【病因与发病机制】
(一)血小板生成障碍或无效生成
1.巨核细胞生成减少
(1)物理、化学因素:
如电离辐射、肿瘤化疗药物、抗生素类(氯霉素、磺胺药)、解热镇痛药(保泰松、吲哚美辛)、抗甲状腺药(丙硫氧嘧啶、甲巯咪唑、卡比马唑)、抗糖尿病药(氯磺丙脲)、抗癫痫药(苯妥英钠)、苯及无机砷等,此类药物干扰DNA合成,抑制细胞丝状分裂,表现为骨髓增生低下和巨核细胞极度减少,常伴有全血细胞减少。另有一些药物如氯噻嗪类、甲苯磺丁脲等抑制巨核细胞生成,血小板生成减少。
(2)骨髓浸润性疾病:
如骨髓转移癌、白血病、骨髓瘤、淋巴瘤、骨髓纤维化等,异常细胞浸润骨髓,造血功能受抑,巨核细胞减少,血小板减少。常伴有其他血细胞质和量的异常。
(3)造血干细胞病变:
如再生障碍性贫血、阵发性睡眠性血红蛋白尿、范可尼贫血、骨髓增生异常综合征,表现为全血细胞减少。
(4)感染性疾病:
如风疹、麻疹、腮腺炎、登革热、艾滋病及某些病原菌引起的败血症等。可能是病原体直接损害巨核细胞,使血小板生成减少。肝炎病毒能直接损害骨髓造血干细胞,使全血细胞减少。获得性单纯无巨核细胞性血小板减少性紫癜,亦与病毒感染、药物、巨核细胞生长因子缺乏引起的巨核细胞生成受抑有关。
(5)血小板生成调控紊乱:
少见,包括血小板生成素(TPO)缺乏和周期性血小板减少症。
(6)遗传性疾病:
如血小板减少伴桡骨缺失(TAR)综合征、Chediak-Higashi综合征、Shwachman-Diamond综合征和先天性无巨核细胞性血小板减少性紫癜等。
2.血小板无效生成
见于维生素B 12 、叶酸缺乏、部分阵发性睡眠性血红蛋白尿及骨髓增生异常综合征等,特征为骨髓巨核细胞数量正常或增多,但血小板产率降低,循环中血小板寿命有不同程度的缩短。
(二)血小板破坏增加或消耗过多
血小板寿命缩短、过早破坏或消耗过多,导致周围血中血小板减少。而骨髓中巨核细胞数正常或代偿增生,伴有成熟障碍。常见的病因如下。
1.免疫性破坏
除ITP外,继发性常见病因有:
(1)药物:
药物作为半抗原与血浆蛋白或血小板蛋白质结合成全抗原,产生相应抗体。药物抗体复合物激活补体,损伤血小板,被单核-巨噬细胞系统吞噬。这类药物有奎宁、奎尼丁、铋剂、金盐、洋地黄毒苷、异烟肼、甲基多巴、肝素以及镇静、安眠、抗惊厥药物等。
(2)某些免疫异常疾病:
包括风湿性疾病如系统性红斑狼疮、结节性多动脉炎等;淋巴增殖性疾病如慢性淋巴细胞白血病、淋巴瘤、骨髓瘤等。
(3)感染相关血小板减少:
常见于病毒及细菌感染,如幽门螺杆菌(Hp)感染、流感、麻疹、水痘、出血热、肝炎、艾滋病、新型布尼亚病毒、伤寒及败血症等。这与病毒抗原-抗体复合物致敏血小板或血中抗血小板抗体水平升高引起血小板破坏过多有关。
(4)同种免疫性血小板减少:
包括输血后紫癜和新生儿同种免疫性血小板减少性紫癜。前者是由于血小板表面特异性抗原(如HPA-1a、HPA-2b HPA-4a、HPA-3a等)阴性患者输入相应抗原阳性血液,产生同种抗血小板抗体,当再次输注相应抗原阳性血液时,体内抗体与输入的抗原结合,引起输入的血小板破坏。患者大多为经产妇或有输血史者,输入后患者出现发热、寒战,大约在1周左右血小板急剧下降,伴有严重出血表现。后者由于母亲对胎儿不相容的血小板抗原产生同种血小板抗体,这种抗体通过胎盘进入胎儿体内引起血小板减少。近半数发生在首次妊娠,常见于患儿母亲是HPA-1a阴性而父亲是阳性者,但引起本病发病的抗原型在不同种族间的差异较大。新生儿出生时可见全身散在性紫癜、瘀斑,病程有自限性,一般持续1~2周,很少超过2~4周,约10%的患儿并发颅内出血死亡。
2.非免疫性破坏
血管炎、人工心脏瓣膜、动脉插管、体外循环、血液透析等,由于血管内膜粗糙,血管内异物或血液流经体外管道时可引起血小板机械破坏,血小板黏附在内膜或异物表面,亦可导致血小板减少。
3.血小板消耗过多
主要见于血栓性微血管病,如弥散性血管内凝血、血栓性血小板减少性紫癜、溶血尿毒综合征均因微血管内弥散性血栓形成使血小板消耗过多,导致血小板减少。
(三)血小板分布异常
各种原因的脾大,包括脾肿瘤、脾充血、脾浸润(戈谢病、尼曼-匹克病)、黑热病及原发性脾大等,肿大的脾可以扣留全血85%的血小板。骨髓巨核细胞正常或增多。
(四)假性血小板减少症
检测血常规常用的抗凝剂如乙二胺四乙酸(EDTA)等,可致部分受检者血小板在体外聚集,出现血小板计数减少的假象,称为EDTA依赖性假性血小板减少症。血涂片可发现血小板明显聚集,更换抗凝剂后血小板恢复正常。
继发性血小板减少往往是综合因素,如感染、药物、肿瘤不仅抑制骨髓造血,同时还有免疫性血小板破坏或分布异常。
【临床表现与实验室检查】
患者有原发病表现或发病前有某种致病因素接触史,轻、中度血小板减少(>50×10 9 /L)可无出血表现,重度血小板减少常有皮肤、黏膜瘀点、紫癜、瘀斑、鼻出血、口腔血疱、黑便、月经过多或术后伤口渗血等,颅内出血是主要的死亡原因。实验室检查除血小板减少外可有束臂试验阳性,出血时间延长,血块退缩不佳,凝血检查正常。骨髓涂片检查十分重要,骨髓中巨核细胞减少提示生成障碍,见于再生障碍性贫血及骨髓浸润性疾病等。巨核细胞正常或增多,但可伴有成熟障碍,见于血小板破坏、消耗过多或分布异常等疾病。疑为HIV感染应查T细胞功能及检测血清HIV抗体。疑为系统性红斑狼疮应查血清抗核抗体、抗Sm抗体、抗ds-DNA抗体等。疑为淋巴瘤应做淋巴结活组织检查。TPO检测和网织血小板计数,对鉴别血小板生成减少还是破坏加速有重要价值。前者血清TPO浓度升高,网织血小板计数正常或减少;后者血清TPO浓度正常,而网织血小板计数增加。
【诊断】
患者有出血症状伴血小板减少,同时有下列征象时应考虑本病:①发病前有服药、电离辐射、妊娠或输血史;②既往有出血史或家族出血史;③伴有发热、畏寒等感染症状;④体检有肝、脾、淋巴结肿大,尤其是明显脾大者;⑤失血不多而贫血较重者;⑥伴有白细胞、红细胞量和质的异常。骨髓涂片或活检,对骨髓病性贫血及再生障碍性贫血的诊断有重要意义。若因脾大做脾切除,脾病理检查可能有助于发现引起血小板减少的病因。
【治疗】
主要针对原发病。出血严重时肾上腺糖皮质激素可以改善症状,必要时输注血小板悬液。免疫性血小板减少皮质激素大多有效,部分患者可行血浆置换治疗。药物性血小板减少应立即停服可疑药物,大多在7~10天血小板恢复正常。感染性血小板减少应积极抗感染治疗,一般在感染控制后2~6周血小板恢复正常,感染引起骨髓抑制者病程迁延较长。对脾功能亢进者,可做脾切除治疗。海绵状血管瘤可采取肿瘤照射或手术切除治疗。
主要参考文献
1.张之南,郝玉书,赵永强,等.血液病学.第2版.北京:人民卫生出版社,2011,1281-1295.
2.Gauer RL,Braun MM.Thrombocytopenia.Am Fam Physician,2012,85(6):612-622.
3.Cines DB,Bussel JB,Liebman HA,et al.The ITP syndrome:pathogenic and clinical diversity.Blood,2009,113(26):6511-6521.
第七节
血栓性血小板减少性紫癜与溶血尿毒综合征
化范例 蔡则骥
血栓性血小板减少性紫癜(thrombotic thrombocytopenic purpura,TTP)是以微血管内广泛血小板血栓形成为特征的血栓性微血管病。临床以血小板减少、微血管病性溶血性贫血、神经精神系统症状、发热和肾损害为主要特征,称为TTP五联征。其与溶血尿毒综合征(hemolytic uremic syndrome,HUS)同属血栓性微血管病(thrombotic microangiopathy,TMA)范畴。
【病因与发病机制】
TTP发病与vWF裂解蛋白酶(vWF-CP)即ADAMTS 13的活性明显下降有关。内皮细胞损伤后释放超大分子vWF多聚体(UL-vWF)进入血浆,诱导血小板活化及聚集。在生理情况下,血浆中vWF-CP可将UL-vWF降解成正常大小的多聚体。当vWF-CP缺乏或活性明显减弱时,UL-vWF不能被有效地降解而在血管内诱导血小板聚集,在末梢动脉、毛细血管内形成广泛的血小板血栓,不但导致血小板的消耗性减少,同时红细胞在通过病变微血管时被阻留、破坏,即出现微血管病性溶血性贫血。TTP可分为遗传性和获得性两类。遗传性TTP或称为Upshaw-Schulman综合征是由于 ADAMTS 13 基因缺陷导致vWF-CP活性下降,为半显性方式遗传。获得性TTP是由于体内存在ADAMTS 13抗体。有些无明确病因,称为原发性TTP,有些则继发于感染、药物、肿瘤、妊娠等,称为继发性TTP。因子V Leiden可能与vWF-CP活性正常的TTP患者发病有关。
【临床表现】
临床上TTP以20~60岁女性多见,起病急骤,进展迅速。主要表现有:①微血管病性溶血:因红细胞受机械性损伤而破碎引起,95%以上患者出现不同程度的贫血,部分伴有黄疸或脾大。②血小板消耗性减少:几乎见于所有患者。③神经精神症状:表现变化不定,初期多为一过性,但可反复发作。整个病程中92%的患者有不同程度的意识障碍和紊乱、头痛、眩晕、惊厥、言语不清、知觉障碍、精神错乱、嗜睡甚至昏迷。以上表现通常称为TTP三联征。④发热:可见于不同病期。⑤肾脏损害:肾血管广泛受累导致肾损害,见于约88%的患者,表现为蛋白尿、镜下血尿和管型尿。重者可发生氮质血症和急性肾衰竭。三联征加上肾脏损害、发热称为TTP五联征。⑥其他方面:如心肌、肺、腹腔内脏器微血管受累,均可引起相应的症状。
【实验室与辅助检查】
血液常规检查血小板明显减少,中至重度贫血,网织红细胞升高,涂片中可见破碎红细胞及有核红细胞,50%的患者白细胞升高。骨髓中红系及巨核系代偿性增生,巨核细胞可伴成熟障碍。凝血检查基本正常,Coombs试验阴性,血浆游离血红蛋白增高。血清乳酸脱氢酶和未结合胆红素增高、结合珠蛋白减少以及肾脏损害表现如血尿素氮、肌酐升高等。血浆vWF-CP活性下降。组织病理学检查可在小动脉、微血管中发现均一性透明血栓,PAS染色阳性。
【诊断与鉴别诊断】
TTP的诊断仍为临床诊断,若患者已具备三联征,临床即可诊断为TTP。血浆vWF-CP活性<5%对诊断TTP具有较高的特异性,但活性正常并不能排除TTP的诊断。鉴别诊断包括其他伴有血小板减少和溶血的疾病如弥散性血管内凝血、溶血尿毒综合征、Evans综合征等。
【治疗】
血浆置换为首选的主要治疗方法,其作为一种替代治疗可使TTP的存活率达85%~90%或更高。在开始治疗的前2天,每天置换1.5个血浆容量(约60ml/kg),以后每天置换1个血浆容量(约40ml/kg),直至血小板计数正常和溶血消失。无条件血浆置换者可行血浆输注,但疗效不及前者。遗传性TTP患者一般体内并不存在vWF-CP抗体,故直接进行血浆输注即可达到治疗目的。在血浆置换治疗的同时应用糖皮质激素直至病情缓解,甲泼尼龙0.75mg/kg或泼尼松1.0mg/kg,12小时给药1次。血浆置换联合利妥昔单抗每周375mg/m 2 连用4~8周可明显减少复发。其他制剂如长春新碱、环磷酰胺、环孢素、丙种球蛋白等在难治性、复发性患者中亦可应用。脾切除去除了扣押和破坏血小板和红细胞的场所,也去除了vWF-CP抗体产生的部位,在部分难治患者中有效。除非出现致命性出血或颅内出血,血小板输注是禁忌的。
【预后】
自血浆置换用于治疗TTP,本病的预后有较大改观,病死率由过去的95%~100%降到10%~20%甚至5%以下,但约20%~50%的患者可复发,复发患者上述治疗依旧有效。
溶血尿毒综合征参见第十七篇第七章第十四节。
主要参考文献
1.中华医学会血液学分会血栓与止血学组.血栓性血小板减少性紫癜诊断与治疗中国专家共识.中华血液学杂志,2012,33(11):983-984.
2.Scully M,Hunt BJ,Benjamin S,et al.Guidelines on the di-agnosis and management of thrombotic thrombocytopenic purpura and other thrombotic microangiopathies.Br J Haematol,2012,158(3):323-335.
3.George JN,Nester CM.Syndromes of Thrombotic Microangiopathy.N Engl J Med,2014,371(7):654-666.
第八节
血小板功能障碍性疾病
化范例 蔡则骥
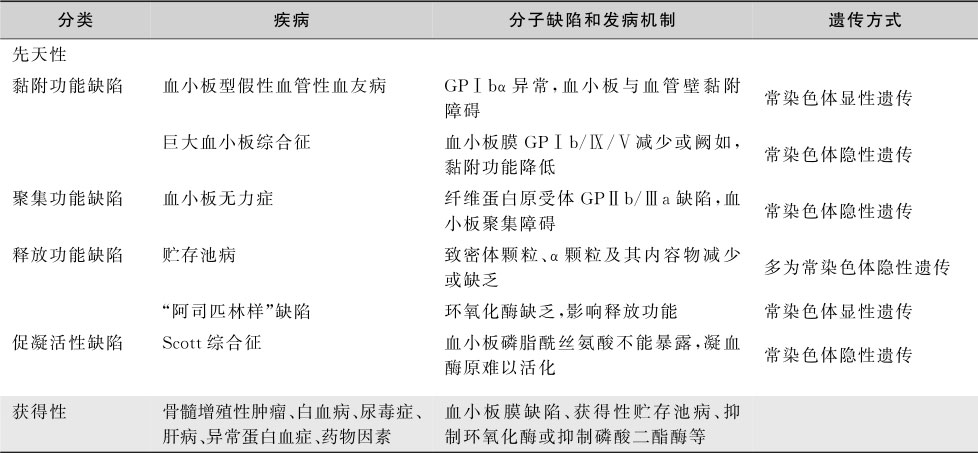
血小板功能障碍性疾病是一组由于血小板结构或代谢异常引起血小板黏附、聚集、释放及凝血活性等功能缺陷所致的出血性疾病,可伴或不伴有血小板计数改变。临床表现为血管-血小板型皮肤、黏膜出血,严重度因病因而异。分为先天性和获得性两大类,见表16-7-5。
表16-7-5 血小板功能障碍性疾病的分类
【先天性血小板功能障碍性疾病】
先天性血小板功能障碍性疾病种类众多,血小板活化过程中涉及的任何膜糖蛋白、受体、信号分子、酶等出现结构或功能的异常均可导致相应的黏附、聚集、释放、促凝等功能障碍。
(一)血小板型血管性血友病
为常染色体显性遗传性疾病。血小板膜GPⅠbα功能异常,使血小板与血浆中vWF多聚体结合反应增强,使血浆中vWF高分子多聚体减少甚至阙如,从而导致血小板黏附缺陷,表现为轻到中度的皮肤、黏膜出血或手术创伤后出血不止。实验室检查包括出血时间延长,血小板计数减少,vWF:Ag含量降低,高分子量vWF多聚体阙如等。治疗上可予输注正常血小板辅以少量冷沉淀物。
(二)巨大血小板综合征(Bernard-Soulier综合征)
为常染色体隐性遗传性疾病。由于血小板膜GPⅠb/Ⅸ/Ⅴ复合物减少或缺乏所致,杂合子型患者无症状,纯合子型婴儿出生后即可有自发性皮肤、黏膜出血甚至内脏出血。血小板计数正常或减少,体积增大似淋巴细胞样,直径可达8μm甚至20μm以上,巨大血小板比例可达30%~80%。出血时间明显延长,凝血酶原消耗不良,血小板对胶原的黏附性降低,对ADP、胶原和肾上腺素的诱导聚集正常,而对瑞斯托霉素、妥布霉素不发生聚集,加入vWF不纠正。确诊依据是GPⅠb/Ⅸ/Ⅴ复合物减少或缺乏。出血严重时输单采血小板有效。非肌性肌球蛋白重链ⅡA异常所致MYH9综合征患者亦具有血小板巨大的特点,但一般不影响血小板功能。
(三)血小板无力症(Glanzmann病)
为常染色体隐性遗传性疾病。血小板膜上GPⅡb/Ⅲa含量减少或缺乏,患者幼年即有皮肤、黏膜甚至胃肠道出血,女性有月经过多,携带者一般无症状。血小板计数和形态正常,对ADP、胶原、肾上腺素和凝血酶诱导的聚集反应减低或阙如,但对瑞斯托霉素的聚集反应正常,出血时间延长,血块退缩不良。血小板第3因子(PF 3 )有效性降低。确诊有赖于GPⅡb/Ⅲa含量检测。
(四)血小板活化受体及信号传导通路异常
血小板活化过程涉及的ADP受体P2Y12、胶原蛋白受体GPVI、TXA 2 受体等异常导致血小板聚集功能缺陷,表现为手术或外伤后轻中度出血。患者血小板对ADP、胶原或肾上腺素等诱导剂的聚集反应减低。
(五)贮存池病
血小板内颗粒缺陷所致的一组出血性疾病,分为致密体颗粒缺陷、α颗粒缺陷以及两者合并缺陷。
1.致密体颗粒缺陷
主要是致密体颗粒内容物ADP、ATP、5-HT等缺乏,使血小板第2相聚集波减弱或消失。临床上可伴有其他遗传性缺陷而形成特定的综合征,如Hermansky-Pudlak综合征中伴有眼及皮肤白化病、蜡样脂褐斑,Chediak-Higashi综合征易发感染,Wiskott-Aldrich综合征则伴有湿疹、免疫缺陷等。
2.α颗粒缺陷
在美蓝-伊红染色下呈灰蓝色,又称灰色血小板综合征,有早期发生骨髓纤维化倾向。单纯α颗粒缺乏少见,常和致密体颗粒同时缺乏。
贮存池病多见于儿童,可有轻到中度皮肤黏膜出血、手术或创伤后过度出血。实验室检查特点有:血小板计数正常或轻、中度减少;出血时间延长;对ADP、肾上腺素第1相聚集波正常而无第2相聚集波;PF 3 的有效性降低。电镜或免疫电镜示血小板和巨核细胞内致密体颗粒或α颗粒减少或缺乏,有助于本病的诊断。
(六)“阿司匹林样”缺陷
血小板致密体颗粒及α颗粒内生物活性物质如 ADP、ATP、5-HT、PF 4 、β-TG等含量正常,但因花生四烯酸代谢障碍包括血小板环氧化酶缺乏、TXA 2 合成酶缺乏等,影响PGG 2 和PGH 2 的形成和TXA 2 的合成,而血小板的释放反应必须有TXA 2 的参与。阿司匹林可抑制环氧化酶,其表现与本病相似,因而得名。患者自幼有出血倾向,也有因外伤或手术出血较多才被发现。实验室检查:血小板数量正常,出血时间轻度延长,血小板对玻珠的黏附正常,对花生四烯酸无聚集反应,其余基本同贮存池病。
(七)Scott综合征
原称血小板第3因子(PF 3 )缺乏症。当血小板被激活时,血小板内膜的磷脂酰丝氨酸不能有效外翻,导致因子Ⅴa和因子Ⅹa无法与血小板结合,凝血酶原不能转化为凝血酶。Ca 2+ 的内流障碍或其下游的移位酶(translocase)活性异常均可影响磷脂酰丝氨酸的翻转从而影响第二阶段的凝血过程。患者在受到外伤或者接受侵袭性操作时容易出血过多。血小板计数、黏附、聚集、释放反应均正常。血清凝血酶原时间缩短,多种诱导剂测定PF 3 有效性均降低。
先天性血小板功能障碍患者应避免剧烈运动、肌内注射、抗血小板药及抗凝药的使用。糖皮质激素和抗纤溶药物可改善出血症状,DDAVP及FⅦa亦可应用,但输注血小板仍是严重出血或外科手术时的主要措施。Bernard-Soulier综合征和Glanzmann病患者由于缺乏相应血小板膜糖蛋白,反复输注血小板可诱发同种免疫而出现无效输注,故应视为紧急措施仅在必要时给予。严重的 Wiskott-Aldrich综合征、Bernard-Soulier综合征、Glanzmann病等患者权衡利弊后可进行骨髓移植或基因治疗。
【获得性血小板功能障碍性疾病】
患者无出血病史及家族出血史,但有导致血小板功能障碍的原发病或服药史,发病机制复杂,发病率远高于先天性血小板功能缺陷性疾病,常影响患者的止血和血栓形成。常见病因如下:
(一)药物
①影响血小板膜的药物:如ADP受体拮抗药噻氯匹定、氯吡格雷,GPⅡb/Ⅲa拮抗药阿昔单抗、替罗非班、依替巴肽,以及青霉素、头孢菌素、右旋糖酐等。上述药物通过阻断血小板膜受体和膜糖蛋白抑制血小板的黏附和聚集功能,部分通过抑制血小板的释放反应。②抑制血小板环氧化酶的药物:如阿司匹林、吲哚美辛、布洛芬、保泰松和苯磺唑酮等,使花生四烯酸不能合成前列环素(PGI 2 )和TXA 2 ,影响血小板的释放功能。③作用于血小板环腺苷系统的药物:如双嘧达莫(潘生丁)、茶碱、咖啡因、前列环素、异丙肾上腺素等,通过抑制磷酸二酯酶或活化腺苷酸环化酶使血小板内cAMP增多,抑制血小板的聚集。④其他:包括肝素、链激酶、尿激酶、t-PA、硝酸甘油、维拉帕米、硝苯地平、速尿、奎尼丁、ACEI制剂、三环类抗抑郁药、吩噻嗪类药物、选择性5-HT再摄取抑制剂、全麻药氟烷、抗肿瘤药物普卡霉素(光神霉素)、柔红霉素以及造影剂等,均可影响血小板的功能,抑制血小板的聚集。
(二)疾病
常见于:①某些血液病:如骨髓增殖性肿瘤(MPN)由于血小板形态及膜糖蛋白异常、致密体和α颗粒及内容物减少(获得性贮存池病),出血及血栓形成是其主要并发症;异常球蛋白血症包括多发性骨髓瘤(MM)、华氏巨球蛋白血症(WM)及意义未明的单克隆丙种球蛋白病(MGUS)等,其出血不仅因血小板减少,还与异常蛋白覆盖于血小板表面,影响血小板黏附、聚集和释放功能有关;急性白血病和骨髓增生异常综合征(MDS),出血的主要原因是血小板减少,部分是由于血小板畸形伴颗粒异常,聚集功能异常及促凝活性降低。②尿毒症患者血浆中积聚的尿素等代谢产物损害血小板的黏附、聚集和释放功能。③其他疾病:如肝病的出血主要与凝血因子合成减少、纤溶亢进、血小板减少及并发弥散性血管内凝血等有关,但也发现其血小板膜上GPⅠb减少,引起血小板功能障碍。系统性红斑狼疮和其他免疫复合物疾病,可能与血浆或血小板上的IgG能抑制血小板聚集有关,提示为抗体介导的血小板功能异常。
治疗原则首要是处理原发病和停服有关药物,严重出血者应输单采血小板。
主要参考文献
1.张之南,郝玉书,赵永强,等.血液病学.第2版.北京:人民卫生出版社,2011,1296-1308.
2.Michelson AD.Platelets.3th ed.Burlington:Academic Press,2013,971-981.
3.Gresele P,Harrison P,Gachet C,et al.Diagnosis of inheri-ted platelet function disorders:guidance from the SSC of the ISTH.J Thromb Haemost,2015,13(2):314-322.
第九节
血友病
王志梅
血友病(hemophilia)是遗传性凝血因子缺乏引起的出血性疾病,典型血友病患者常自幼年发病、自发或轻度外伤后出血,在外伤、手术时常出血不止。根据缺乏的凝血因子不同,可分为血友病A(FⅧ缺乏症)和血友病B(FⅨ缺乏症)。在凝血酶促反应过程中,FⅧa作为FⅨa的非酶性辅因子,在Ca 2+ 及磷脂存在的条件下,激活FⅩ生成凝血活酶。血友病A或B由于FⅧ或FⅨ促凝活性减少导致凝血活酶生成障碍,凝血时间延长。欧美各国发病率约为5~10/10万人口,日本10.4/10万人口,据1992年我国24个省市37个地区普查其发病率为2.72/10万人口,其中血友病A约占80%。
【病因与发病机制】
FⅧ 、 FⅨ 基因位于X染色体上,血友病A与血友病B均表现为伴性隐性遗传特征,即女性传递,男性发病。若男性血友病患者与正常女性结婚,其女儿均为携带者,儿子则全部健康;若女性血友病携带者与正常男性结婚,其儿子半数发病,女儿也有半数为携带者;若男性患者与女性血友病携带者结婚,其儿子与女儿均有半数发病的概率,女儿还有半数为携带者;若男性患者与女性患者结婚,其下一代不论男女均为血友病患者。约70%的血友病A患者有阳性家族史,30%的病例无家族史,其中部分可能是由于基因突变所致。FⅧ基因位于染色体Xq28,FⅧ的合成部位未完全阐明,但肝脏间质细胞、外周血细胞及某些淋巴细胞都有FⅧ基因的表达。血友病A的基因缺陷类型主要包括内含子22倒位、FⅧ基因点突变、缺失或有异常基因插入等,基因缺陷致使FⅧ合成障碍或FⅧ分子结构异常导致促凝活性降低。FⅧ在正常人血浆中的含量仅为0.1mg/L,是所有凝血因子中含量最低的。FⅧ在血液循环中与其载体血管性血友病因子(vWF)以复合物形式存在。FⅧ的生物活性通过FⅧ促凝成分(FⅧ∶C)来实现,FⅧ∶C的正常值为50%~150%。劳动、剧烈运动、注入肾上腺素和应激状态,均能使血浆FⅧ∶C水平增高。FⅧ∶C不稳定,4℃存放24小时后活性下降20%;FⅧ∶C输入体内后半衰期仅12小时。血友病B占血友病总数的15%~20%,其基因位于染色体Xq26.3-27.2。FⅨ在肝内合成,是维生素K依赖性凝血因子。血浆FⅨ水平为10mg/L,是FⅧ的100倍。
【临床表现】
主要表现为异常出血及出血所致压迫症状或并发症。肌肉关节腔或深部组织出血、创伤后过量出血是本病的特征性表现。肌肉出血多见于负重的肌肉群,如腰大肌、腿部、臀部等,可形成血肿,局部肿痛,活动受限。关节出血多累及负重或活动较多的大关节,如膝关节,其次为踝、髋、肘、腕及肩等关节。急性期因关节腔及周围软组织出血致使局部红肿疼痛,活动受限。多数患者因反复关节腔出血致使血液不能完全吸收,形成慢性炎症,滑膜增厚、纤维化,软骨变性及坏死,最终关节僵硬、畸形变,周围肌肉萎缩,导致正常活动受限。消化道出血、血尿也较为常见。颅内出血及硬脊膜下血肿不常见,多发生于外伤后,病死率高。深部组织内血肿可压迫附近血管引起组织坏死,压迫神经可产生疼痛、麻痹等症状。口腔、喉、舌或颈部严重出血可引起窒息。皮肤、黏膜出血并非血友病的特征性表现,但由于皮下组织、口腔黏膜、牙龈、舌等部位容易受伤,故损伤后过量出血常见。创伤后异常出血也是血友病的主要表现,如拔牙、肌内注射等可导致持久的出血或渗血,可历时数天甚至数周。血友病B重型患者的比例较血友病A少,FⅨ水平明显低于正常者,有出血表现。
【临床分型】
(一)血友病A的临床分型标准
1.重型
约50%~60%的患者为重型患者,其血浆中FⅧ∶C<2%,常在2岁以前就有严重出血,甚至结扎脐带时出血不止。
2.中间型
FⅧ∶C为2%~5%,约占患者总数的25%~30%,起病在童年时期以后,以皮下及肌肉出血居多,亦有关节腔出血。
3.轻型
约占15%~20%,FⅧ∶C为5%~25%,出血多发生在青年期,由于运动、拔牙或外科手术后出血不止而被发现,出血轻微,可以正常生活。
4.亚临床型
FⅧ活性为25%~40%,通常只在大手术后才发生异常出血。
(二)血友病B的临床分型标准
1.重型
FⅨ∶C<2%。
2.中间型
FⅨ∶C 2%~5%。
3.轻型
FⅨ∶C 5%~40%。
以上各型临床表现与同型血友病A相仿。
【实验室检查】
(一)筛选试验
出血时间、血小板计数、凝血酶原时间测定均正常,活化部分凝血活酶时间(APTT)和硅化凝血时间(SCT)在血友病是可能会出现异常。APTT可以检测出FⅧ∶C<25%的患者,SCT可以检测出FⅧ∶C 25%~45%的患者。
(二)纠正试验
常用凝血活酶生成试验Biggs法(BTGT)、简易凝血活酶生成试验(STGT)作为血友病的纠正试验。正常血浆经硫酸钡吸附后尚含有FⅧ、FⅪ,正常血清中含有FⅨ、FⅪ,因此,如果患者STGT仅被硫酸钡吸附正常血浆纠正时,为FⅧ缺乏症;仅被正常血清纠正时,为FⅨ缺乏症;如两者皆可纠正,则为FⅪ缺乏症。借此可将三种凝血因子缺乏加以鉴别。
(三)确诊试验
需进行FⅧ∶C和FⅧ∶Ag测定、FⅨ∶C和FⅨ∶Ag测定。在多数血友病患者中,血浆抗原水平与活性水平平行减少,但部分患者的抗原与活性水平不平行。通过检测FⅧ抗原水平,将血友病A分为三类:血浆中无法检测到FⅧ抗原,为交叉反应物质阴性型(CRM-);如果血浆中可以检测到抗原,且抗原和活性平行降低则称为交叉反应物质下降型(CRMred);另有约5%的患者血浆中可以检测到抗原,但活性降低程度大于抗原,称为交叉反应物质阳性型(CRM + )。CRM-者占血友病A的3/4左右,此类患者接受替代治疗后易于产生抑制性抗体;CRM + 占5%,此类患者的发病主要是FⅧ∶C结构和功能异常。90%的血友病B为CRM - ,10%为CRM + 。
【诊断标准】
1.多为男性患者,有或无家族史。
2.有肌肉、关节腔或深部组织出血或手术创伤后过量出血的表现。
3.实验室检查PT正常,APTT延长,亚临床型可正常,血小板计数、出血时间、凝血酶原时间正常,FⅧ∶C或FⅨ∶C减少。
4.排除继发性相应凝血因子减少。
【鉴别诊断】
(一)血管性血友病
为常染色体遗传性疾病,两性均可发病;出血好发于黏膜和内脏,很少累及关节腔及肌肉深部,罕见关节畸形,随着年龄增长出血症状减轻;实验室检查发现出血时间延长,血小板黏附率降低,多数患者的血小板对瑞斯托霉素无凝集反应,血浆中FⅧ∶C/vWF∶Ag比例增高或正常,血浆中vWF减少或结构异常。
(二)FⅪ缺乏症
本病为常染色体隐性遗传疾病,两性均可发病,自发性出血不多见,检测FⅪ∶C可诊断。
(三)获得性血友病
非血友病患者血液中存在FⅧ或FⅨ抑制物。患者多为70岁以上老年人,其余患者多数有自身免疫性疾病基础。获得性血友病患者血液中可检测到FⅧ抗体,与血友病A鉴别应不难。
FⅧ或FⅨ减少还可见于严重肝脏疾病、严重胆道梗阻、抗凝治疗、长期广谱抗生素治疗、弥散性血管内凝血等疾病状态。
【治疗】
(一)预防
1.加强宣教 将疾病的性质、防治知识向患者、家属、学校及单位宣教,避免创伤及剧烈活动,鼓励适当体力活动。
2.避免使用抗血小板药物。
3.避免肌内或皮下注射,静脉穿刺后至少压迫5分钟以预防出血。
4.尽量避免手术,如必须施行手术,应在术前补充所缺乏的凝血因子。
5.有条件者应定期预防性补充相应凝血因子等。美国国家血友病基金会的医学专家委员会推荐对重型血友病患者进行预防性治疗,维持FⅧ∶C或FⅨ∶C>1%。推荐剂量为:每次FⅧ25~40U/kg,每周3次,每次FⅨ25~40U/kg,每周2次。
6.对血友病患者家人特别是女性患者,应做基因检测,产前诊断,进行优生优育。
(二)局部止血治疗
若发生轻微损伤,可用吸收性明胶海绵、纤维蛋白泡沫、凝血酶、肾上腺素等局部压迫止血。国外配制止血剂中含冷沉淀、凝血酶、氨基己酸,用于血友病A患者的局部治疗止血效果较好。
(三)替代治疗
1.新鲜血浆和新鲜冰冻血浆
含所有的凝血因子,每1ml新鲜血浆内含1U FⅧ∶C和1U FⅨ∶C;血友病B患者可采用储存血浆治疗 (以5天内为宜)。一次最大安全量为10~15ml/kg,由于容量因素限制,单纯输注新鲜血浆和新鲜冰冻血浆难以使FⅧ∶C或1U FⅨ∶C达到有效止血浓度。
2.冷沉淀制剂
每袋20ml冷沉淀制剂取自200ml新鲜血浆,约含有80~100U FⅧ,适用于轻型和中型血友病A患者。冰冻保存于-20℃以下,室温下放置1小时活性将丧失50%。
3.凝血酶原复合物浓缩剂 (PCC)
每瓶200U,相当于200ml血浆中所含有的因子Ⅸ,适用于血友病B。
4.FⅧ浓缩剂
是从多份冷沉淀制剂中提取和制备得来,已灭活病毒。
5.重组FⅧ及FⅨ(rFⅧ及rFⅨ)
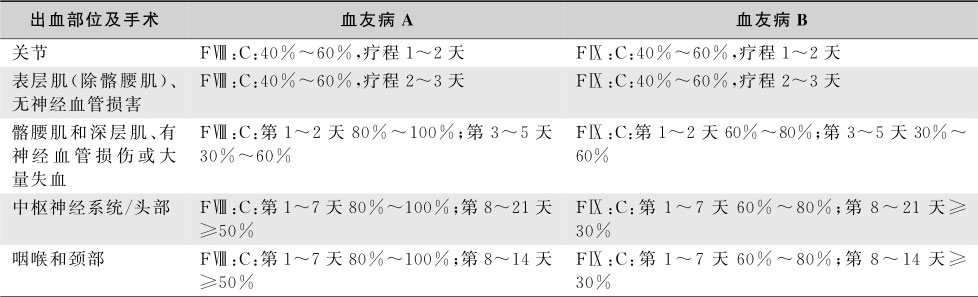
rFⅧ的功能特征、药代动力学等生化特征与血浆源性制品大致相同,临床应用安全、有效,少数患者应用后可产生FⅧ抑制性抗体但多数滴度不高,对诱导免疫耐受治疗反应良好。第一代rFⅧ为全长FⅧ,利用人体白蛋白作为稳定剂。第二代rFⅧ利用蔗糖代替蛋白作为稳定剂,使病毒传播性疾病的发生率进一步降低。替代治疗的目标是将患者血浆FⅧ、FⅨ活性水平提高到足以止血的水平。每千克体重输入1U FⅧ,可使体内FⅧ的活性升高2%。FⅧ在体内的生物半衰期仅12小时,需12~24小时输注1次,病情严重者8~12小时1次。血友病A患者轻度出血时,通常需要将FⅧ水平提高至正常人的25%~30%,重度出血需提高至50%以上。每输注1U FⅨ提高其活性0.5%~1%。推荐rFⅨ用量=体重(kg)×预期增加值(%)×1.2。FⅨ首剂半衰期约2~3小时,以后为20~30小时,故第一次输注后2~4小时就应做第二次输注,以后每24小时输注1次。按照血友病诊断与治疗中国专家共识(2013年版),不同疾病状态下替代治疗方法参照表16-7-6和表16-7-7。
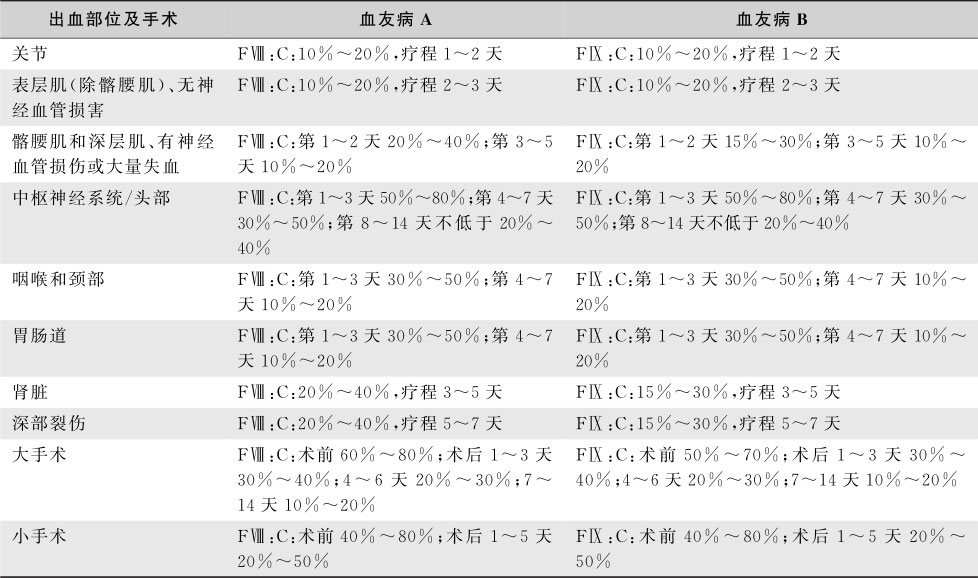
表16-7-6 血友病患者获取凝血因子不受限时的替代治疗方案(反应不充分时疗程可适当延长)
续表
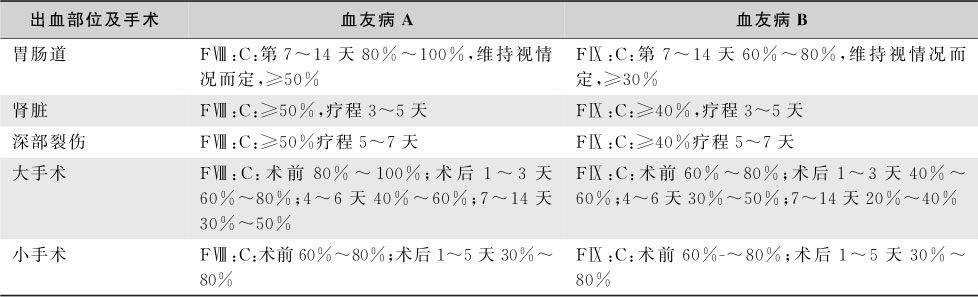
表16-7-7 血友病患者获取凝血因子受限时的替代治疗方案
(四)血友病患者的外科手术问题
应尽量避免。术前数小时应开始补充凝血因子使之正常,术中可维持静滴,术后监测至少2次/天,使谷浓度达到足够止血的水平,替代治疗持续到创口完全愈合。膝关节、髋关节置换时替代治疗需数周。
(五)DDAVP(1-去氨基-8-右旋精氨酸加压素,1-desamino-8D-arginine vasopressin)
是一种人工合成的抗利尿激素衍生物,有增加血浆内FⅧ水平的作用,静脉注射后可使FⅧ∶C及vWF∶Ag增加2~3倍。适用于轻型血友病A和血友病A携带者。每次剂量为0.3~0.5μg/kg,通常每次剂量不超过20μg。缓慢静脉注射,注射后30~60分钟作用达高峰。因该药能激活纤溶系统,需同时合用抗纤溶药物。每12小时1次,2~5天为一疗程。DDAVP也可鼻腔喷雾使用,剂量须提高到静脉用量的10倍左右。副作用包括暂时性面部潮红及水滞留,水滞留作用可持续24小时。
(六)其他药物治疗
1.抑制纤维蛋白溶解药物
可保护已形成的血凝块不易被溶解,与替代疗法同时合用有协同作用;部分轻型血友病患者在口腔小手术时单独应用疗效满意。常用药物有:6-氨基己酸0.1g/kg,每日口服3~4次,或4~6g溶于100ml 5%葡萄糖液或盐水中静滴;氨甲环酸250mg每日口服3~4次,或250~500mg每天1~2次静滴。肾出血不宜应用,以免造成梗阻。
2.达那唑 (danazol,炔羟雄烯异唑)
是一种合成的雄性激素,每日400~600mg可提高Ⅷ因子活性水平。
3.女性避孕药复方炔诺酮
每日1mg,连用1~2个月,可提高Ⅷ因子浓度,对血尿、深部组织血肿有一定疗效。
4.糖皮质激素
对减少出血、促进急性积血吸收、减少局部炎症反应均有一定的疗效。
(七)基因治疗
血友病是单基因病,病因明确;凝血因子因可在多种细胞中合成,所以靶细胞选择余地大、治疗效果直观。临床基因治疗的策略是在患者体细胞中增加一个有功能的外源基因,并通过这个基因的表达产物来弥补生理缺陷,以达到治疗目的。
(八)替代治疗的不良反应及处理
1.产生抑制性抗体
是血友病治疗最主要的并发症。血友病A患者反复输注FⅧ后数周至数月内可产生抗FⅧ抗体,有文献报道发生比例可高达30%,尤其好发于婴儿及儿童。该抗体属IgG,对FⅧ∶C有特异性中和作用。血友病B产生抗FⅨ抗体者较少,仅占1%左右。抑制性抗体使输注的凝血因子很快被中和,给治疗带来很大困难。对这些患者治疗的目的是止血、去除抗体。①急性出血时的治疗方法:轻中度出血:无论抗体滴度水平如何,推荐使用FⅧ旁路途径药物治疗。FⅧ旁路途径药物有两种选择:a.rFⅦa:rFⅦa理论上能在组织损伤部位发挥止血作用,同时可以避免血栓形成事件。单独使用rFⅦa治疗8小时以内关节腔出血、一般外科手术中急性出血、有抗体生成的血友病患者择期手术,可取得理想的疗效。通常剂量为90~110μg/kg,每2~3小时1次,24小时后间隔时间可延长,关节腔出血和一般外科手术中急性出血平均注射2~3次可达止血,较大的手术需给药3~5天或直至创伤愈合。b.凝血酶原复合物浓缩剂(PCC)和活化凝血酶原复合物浓缩剂(APCC),对存在抗体的血友病者治疗有效率为50%~60%。需注意制剂中含有少量活化的凝血因子,可能诱发血栓形成。严重出血时:FⅧ抗体有相对的种属特异性,当抗体为抗人FⅧ抗体时,输注高纯度的猪FⅧ制剂能有效止血。但反复输注会产生抗猪FⅧ抗体,少部分患者有发热、血小板减少。抑制性抗体滴度<10Bu时FⅧ(或FⅨ)60~100U/kg每8~12小时1次直到出血被控制,或使血浆FⅧ∶C或FⅨ∶C水平达60%~100%;输后抗体显著升高的患者推荐联合免疫抑制剂如环磷酰胺、肾上腺糖皮质激素治疗。抑制性抗体滴度>10Bu或抗猪FⅧ抗体滴度高时,即使输注大剂量FⅧ或FⅨ也很难起效,血浆置换或体外抗体吸附联合大剂量FⅧ或FⅨ及免疫抑制治疗有效,但急性严重出血时患者血流动力学稳定与否、血浆置换及体外抗体吸附需要特殊的设备及专业人员,限制了其应用。②免疫耐受诱导(ITI)治疗:诱导免疫耐受去除抗体,输注大剂量凝血因子联合免疫抑制治疗可抑制抗体生成效应,反复治疗后可诱发部分免疫耐受性,使高反应者转成低反应者。目前临床多在输入FⅧ或FⅨ后继以大剂量静脉丙种球蛋白、环磷酰胺,规则地输注FⅧ或FⅨ,多数患者可在2~3周内产生免疫耐受,抗体滴度下降,但FⅨ抗体阳性者输入大剂量FⅨ浓缩剂有诱发超敏反应的危险。国内推荐的一线方案为糖皮质激素单用[泼尼松1.0 mg/(kg·d)]或者联合环磷酰胺[1.5~2.0 mg/(kg·d)]。若治疗4~6周后无反应,应该考虑换用利妥昔单抗单用或者联合糖皮质激素的替代治疗方案
2.病毒感染
输注血制品存在肝炎病毒、HIV等感染的可能,重组凝血因子在很大程度上避免了病毒传播的可能。
3.血栓形成事件
大剂量使用PCC等制剂时,因其中含有部分已活化的凝血因子,治疗中有发生血栓形成的可能。
4.溶血
FⅧ浓缩剂是从大量混合血浆中制备而来,其中混有抗A、抗B同型凝集素,大量输注FⅧ浓缩剂有发生溶血的可能。反复多次输注FⅧ,患者体内结合珠蛋白水平较低,可能是因为体内存在慢性亚临床型溶血。
主要参考文献
1.张之南,郝玉书,赵永强,等.血液病学.第2版.北京:人民卫生出版社,2011.
2.Mannucci PM,Mancuso ME,Santagostino E.How we choose factor Ⅷ to treat hemophilia.Blood,2012,119(18):4108-4114.
3.中华医学会血液学分会血栓与止血学组,中国血友病协作组.血友病诊断与治疗中国专家共识(2013年版).中华血液学杂志,2013,34(5):461-463.
第十节
血管性血友病
王志梅
血管性血友病(von Willebrand disease,vWD)于1926年由Eric von Willebrand首先报道,是最多见的遗传性出血性疾病,其发病率约占人口的1%,但出现临床症状者仅 占患者的0.1%。血管性血友病因子(von Willebrand factor,vWF)具备稳定FⅧ活性以及介导血小板与内皮间的黏附作用,在止血中发挥着至关重要的作用。因vWF发生量或质的改变而导致止血功能缺陷,即称为vWD。临床上以皮肤、黏膜出血为主要表现,出血时间延长。本病为常染色体遗传性疾病,多数患者为显性遗传,少数为隐性遗传。
【病因与发病机制】
vWF基因位于12号染色体短臂末端,vWF由血管内皮细胞和巨核细胞合成,血小板中也含有vWF。正常人血浆中的vWF由不同数量亚单位、分子量变化范围很大的多种多聚体组成。血管内皮细胞受损时,内皮下组织中的vWF暴露于血液中,血小板通过GPⅠb与vWF结合黏附于血管内皮下组织,血小板活化后GPⅡb/Ⅲa进一步与vWF结合,最终形成血小板血栓;vWF作为瑞斯托霉素辅因子,加速瑞斯托霉素诱导的血小板聚集。vWF含量减少或与GPⅠb相互作用处分子结构改变,将影响血小板黏附于受损血管,出血时间延长。vWF的另一个重要功能是与FⅧ非共价结合,稳定FⅧ∶C,间接影响凝血过程。vWF含量减少或与FⅧ结合处分子结构改变将导致FⅧ∶C灭活加速,出现二期止血障碍。vWF基因异常,使vWF的合成或释放减少、多聚体形成障碍或出现vWF质的异常,导致vWD的发生。
【临床表现】
本病的出血倾向差异很大,随年龄增长出血症状可减轻。出血多见于皮肤和黏膜,鼻腔和齿龈出血、月经过多是最多见的表现,拔牙及创伤后过度出血也常见,亦可有消化道出血、血尿等。根据病理生理学改变,临床上将vWD分为三型:1型最多见,约占vWD的3/4,vWF多聚体浓度降低而多聚体的结构和功能正常,为常染色体显性遗传,临床上有轻到中度出血症状。2型vWF中大多数缺乏较大分子量的多聚体并伴有功能异常,多为常染色体显性遗传。高分子量vWF多聚体缺失且与血小板结合能力明显降低者为2 A型,约占2型的75%;与血小板GPⅠb结合明显增加者为2B型,约占2型的20%,此型的重要特点为血小板减少、缺乏大的多聚体,vWF可自发地或在低浓度瑞斯托霉素时与血小板结合;多聚体分布正常,vWF质的异常使之与血小板GPⅠb亲和力降低者为2 M型;vWF量与多聚体分布正常,但vWF质的异常导致与FⅧ亲和力降低者为2N型,出血表现较重。3型vWF几乎完全缺失,FⅧ∶C<10%,甚至<1%,终身出血症状严重,可发生自发性关节和肌肉出血而致残。为常染色体隐性遗传,发病率最低。
【实验室检查】
出血时间(BT)延长为本病的主要特点,但部分患者可正常。血小板黏附率减低,除2B型外多数患者对瑞斯托霉素诱导的聚集反应下降,重者几乎不发生聚集,而对其他诱导剂反应正常。2B型及血小板型血管性血友病之血小板可对低浓度瑞斯托霉素起聚集反应。FⅧ∶C和FⅧ抗原含量正常或减低。通过对vWF抗原的定量测定、琼脂糖凝胶电泳多聚体分析,及瑞斯托霉素辅因子活性测定等检查进行临床分型。
【诊断与鉴别诊断】
(一)本病的诊断要点
自幼有皮肤、黏膜出血史,症状随年龄增长而减轻;约半数病例有家族史;出血时间延长;血小板黏附率降低及对瑞斯托霉素聚集反应减弱或不聚集;FⅧ∶C正常或降低;vWF抗原减少或多聚体分布或功能异常。
(二)鉴别诊断
1.血友病A
血友病A为X性联遗传性疾病,实验室检查的主要特征为FⅧ减少,有关vWF检查无异常。
2.巨大血小板综合征
其生化特点为血小板GPⅠb缺乏,导致出血时间延长;血涂片可见特征性的巨大血小板,vWF功能正常。
3.血小板型血管性血友病
又称“假性”vWD。是血小板膜糖蛋白缺陷,与vWF亲和力增高,使血浆中vWF浓度降低引起类似于vWD的表现,部分患者血小板减少,vWF功能正常。
4.获得性血管性血友病
又称vWD综合征。本病的临床表现类似于遗传性血管性血友病。发病是由于vWF合成减少、消耗增加或抗体产生所致。常见于淋巴细胞增殖性疾病、慢性骨髓增殖性疾病、心血管疾病、免疫性疾病和其他恶性肿瘤等,治疗以原发病为主,必要时可静脉输注大剂量免疫球蛋白或替代治疗。
【治疗】
轻型患者可不需特殊治疗,但禁服阿司匹林、双嘧达莫、吲哚美辛、前列腺素E等影响血小板功能的药物。尽量避免手术,必须手术时应作好充分准备。口服避孕药如复方炔诺酮等,可使月经过多及持续时间延长的症状改善。治疗目的是使BT、FⅧ∶C恢复正常,以往成功的治疗经验有重要的参考价值。
(一)DDAVP
可促使内皮细胞中vWF释放入血,FⅧ∶C增高。DDAVP对大多数1型和2型患者有效,对3型VWD无效。应用剂量、方法及注意事项见本章第九节“血友病”。2B型不宜使用DDAVP治疗。
(二)替代治疗
3型及对DDAVP反应差者在出血或围术期需替代治疗。输注冷沉淀物可使BT和凝血异常同时得到纠正,为首选制剂,但其制备过程中无病毒灭活过程;新鲜冷冻血浆也可使用;高纯度FⅧ制剂中缺少与vWF活性有关的高分子多聚体,不能纠正BT延长,不宜选用。输注正常血浆10~15ml/kg可使血中FⅧ∶C活性达75%以上,同时能纠正出血时间延长。vWF半衰期为24小时,故必要时应每24小时或48小时使用输血浆1次,每次5ml/kg。术前输400~600ml血浆,可使出血时间维持在2~4分钟内。
(三)其他治疗
纤溶抑制剂如6-氨基己酸(EACA)和氨甲环酸对月经过多者有一定疗效,对常规剂量无效的月经过多患者,氨甲环酸口服剂量可达到3g/d。某些非损伤性运动可提高FⅧ∶C及vWF浓度,也有缩短出血时间的作用。
主要参考文献
1.中华医学会血液学分会血栓与止血学组,中国血友病协作组.获得性血友病A诊断与治疗中国专家共识(2014年版).中华血液学杂志,2014,35(6):575-576.
2.Xu J,Yu Z,Zhang L,et al.Diagnosis and management of Von Willebrand's disease in China.Semin Thromb Hemost,2011,37:607-614.
3.James PD,Lillicrap D.vonWillebranddisease:clinical and la-boratory lessons learned from the large von Willebranddisease stud-ies.Am J Hematol,2012,87:s4-11.
第十一节
其他遗传性凝血因子缺乏症
王志梅
一、遗传性凝血因子Ⅺ缺乏症
遗传性凝血因子Ⅺ(FⅪ)缺乏症为常染色体隐性遗传性疾病,临床少见。FⅪ又称凝血活酶前质,是丝氨酸蛋白酶,可被FⅫa和凝血酶激活,FⅪa可激活FⅨ,这在内源性凝血途径中对维持凝血酶的不断产生十分重要。杂合子患者血浆中FⅪ水平多在正常人的25%以上,有的甚至高达70%,纯合子患者的FⅪ水平多在15%以下,同时合并FⅪ功能异常者少见。临床上纯合子有出血倾向,杂合子若不合并其他凝血功能障碍应无症状。手术或外伤后出血是最主要的表现,出血可发生在创伤即刻或数小时、数天之后。自发性出血如鼻及牙龈出血、皮肤瘀斑、月经过多等少见。出血倾向与FⅪ含量并不呈正相关,患者既往出血史或其家属出血表现对判断病情的严重程度更有价值。FⅪ缺乏症常合并其他先天性凝血因子异常,如合并FⅤ、Ⅶ缺乏症。实验室检查重型者CT、APTT正常或延长,B-TGT生成不佳,正常吸附血浆和正常血清均可纠正;FⅪ活性及抗原测定具有诊断价值。轻型患者不需治疗,重型患者必须手术者应作好术前准备。通常需将FⅪ水平升至30%以上,大手术、手术部位纤溶活性高时需将FⅪ水平升至45%。FⅪ的生物半衰期为2天左右,在4℃以下活性稳定,故替代治疗时可用贮存血浆。输注血浆7~20ml/kg,可使FⅪ水平提高到25%~50%。手术前输血浆30ml/kg,以后每天5ml/kg或隔日10ml/kg,维持至伤口愈合。冷沉淀物的上层液中约含FⅪ1000U/L,也可用于替代治疗。
二、遗传性纤维蛋白原缺乏症
遗传性纤维蛋白原缺乏症为常染色体显性或隐性遗传性疾病,临床较少见,男女均可发病,其中半数病例有近亲婚配史。纤维蛋白原由肝细胞合成及分泌。根据纤维蛋白原减少的程度,将本病分为无纤维蛋白原血症、低纤维蛋白原血症两种。正常人血浆纤维蛋白原含量为2~4g/L,引起出血的临界水平为0.6g/L。血浆纤维蛋白原含量<0.4g/L称为无纤维蛋白原血症,0.5~0.8g/L称为低纤维蛋白原血症,部分可伴有异常纤维蛋白原血症。无纤维蛋白原血症患者终身具有出血素质,约半数患者出生时脐带出血不止,大片瘀斑、突发胃肠道出血尤为常见,女性可有月经过多,20%的患者有关节腔出血。出生时颅内出血、轻微创伤后出血是致死的常见原因。多数低纤维蛋白原血症患者无出血症状,轻度自发性出血、外伤或术后严重出血是其常见表现。两种患者临床上常见创面愈合延迟和不佳,感染不易局限。所有的凝血检查均可异常,CT、PT、APTT、TT均可延长甚至不凝,但正常血浆或正常纤维蛋白原均可以纠正。纤维蛋白原严重缺乏时出血时间可延长,血小板凝聚率降低。纤维蛋白原在体内的生物半衰期为3~5天,故替代治疗应每周2次。纤维蛋白原制剂开始45~50ml/kg,以后20~25ml/kg,足以达到止血效果。
三、遗传性异常纤维蛋白原血症
遗传性异常纤维蛋白原血症是一种常染色体遗传性疾病,其纤维蛋白原结构和功能异常,能连续几代发病,可为杂合子或纯合子。女性患病较多见。异常纤维蛋白原的命名以首次发现的地区命名。发病机制以单碱基突变为主,并通过影响纤维蛋白肽链释放、纤维蛋白单体聚合、纤维蛋白多聚体交联等途径影响纤维蛋白原发挥正常功能。本病患者的临床表现主要是出血倾向、血栓形成或伤口愈合不佳,也有无症状者。若异常纤维蛋白单体不能进行共价交联,则所形成的纤维蛋白凝块疏松、不坚固,以致伤口愈合延迟、裂开或不良。实验室检查凝血酶时间延长,钙离子可使其部分纠正。纤维蛋白原含量用免疫法测定结果正常,而用凝固法测定可有中等度降低。治疗主要依据临床上是否有出血,如有急性出血或外科手术可考虑输注血浆或纤维蛋白原制剂。
四、遗传性凝血酶原缺乏症和遗传性异常凝血酶原血症
两者均极为罕见,为常染色体隐性遗传,临床表现相似,有轻到中度皮肤黏膜及软组织出血。FⅡ∶C<1%时有自发性出血及创伤后出血,术后或创伤后出血明显,鼻腔及牙龈出血、皮下出血血肿、月经过多常见,关节腔出血很少见。FⅡ∶C在2%~5%时出血表现差异很大,可无症状,也可表现为轻微创伤后严重出血。FⅡ∶C在5%~50%者多无出血症状或仅在大手术或严重创伤后出血。实验室检查示PT延长,APTT延长,TT正常,FⅡ∶C及FⅡ∶Ag检测有确诊价值。轻度表浅出血通常无须替代治疗,严重出血时或手术前需替代治疗,凝血酶原的有效止血浓度在40%~50%。可选用新鲜冰冻血浆、4℃保存血浆、PCC及FⅡ浓缩剂。手术前输PCC 40U/kg,因FⅡ的半衰期为3天,多数患者给药一次即可,需注意大剂量输注PCC有血栓形成的可能。
五、遗传性凝血因子Ⅴ缺乏症
本病系常染色体隐性遗传,甚少见,发病率不超过1/100万。少数患者父母为近亲婚配。部分患者可合并其他先天性异常,如输尿管畸形、动脉导管未闭等。FⅤ由肝脏合成,半衰期为12~36小时。在生理条件下,FⅤ具有促凝、抗凝双重作用。在凝血过程中,其活化形式FⅤa与Ca 2+ 、磷脂和FⅩa形成的复合物是凝血酶原最主要的激活物,是凝血过程中十分重要的辅因子。另外,蛋白C可以将FⅤ水解为具有抗凝活性而缺乏促凝活性的分子(FⅤac),FⅤac可以作为蛋白C的辅因子,在蛋白S的协同作用下灭活FⅧ∶C等凝血因子,参与抗凝。因此FⅤ基因缺陷可能导致出血或血栓,形成两种完全相反的表型。临床上遗传性FⅤ缺陷症约25%的患者有出血表现,主要有皮肤瘀斑、鼻出血和月经过多、术后出血等,发生在胃肠道及中枢神经系统可危及生命的出血罕见,FⅤ缺陷症患者的出血严重性与FⅤ∶C水平没有一致性,杂合子型FⅤ缺陷症病例往往无明显的临床出血症状,多数纯合子型病例的临床出血症状较轻;部分患者有静脉血栓形成事件。实验室检查将遗传性FⅤ缺陷症分为两型:Ⅰ型为交叉反应物质阴性(CRM - );Ⅱ型是为交叉反应物质阳性(CRM + )。目前报道的病例中绝大多数为Ⅰ型。纯合子可有PT延长、APTT延长,蛇毒及正常血清不能纠正凝血缺陷,但能被硫酸钡吸附正常的新鲜血浆纠正。约1/3的患者BT延长。FⅤ∶C降低具有确诊价值。有严重活动性出血时应予以替代治疗,但正常止血所需的FⅤ∶C水平目前不肯定,一般认为25%即可。先天性FⅤ缺乏者手术前应输注新鲜冷冻血浆,首剂为15~25ml/kg,术后15~25ml/kg,5~10天。
六、遗传性凝血因子Ⅶ缺乏症
本病为少见的常染色体隐性遗传性疾病,部分患者父母为近亲婚配。FⅦ被激活后与FⅢ形成复合物可激活FⅩ、FⅨ,启动凝血过程。临床出血症状与FⅦ∶C水平不成比例。杂合子患者一般无出血症状;纯合子患者若FⅦ<1%,则临床出血表现与重型血友病相似,关节出血致残、致死的颅内出血并不少见。实验室检查特征性表现为PT延长,能被正常血浆或血清纠正;APTT、TT、蛇毒时间测定正常,FⅦ凝血活性(Ⅶ∶C)和FⅦ抗原(Ⅶ∶Ag)测定最有价值。轻度出血可给予抗纤溶治疗,严重出血时rFⅦ为首选制剂,亦可用新鲜冰冻血浆、PCC,一般认为血浆中FⅦ的水平提高到正常的25%就可达到止血目的。
七、遗传性凝血因子Ⅹ缺乏症
本病为常染色体隐性遗传,约50%的患者父母为近亲婚配。出血程度与因子Ⅹ的活性相关。杂合子FⅩ∶C通常高于正常的10%,一般无出血症状;纯合子FⅩ∶C常低于正常的1%,多有出血症状,脐带出血为其早期表现,其他有皮肤黏膜出血及创伤后出血,偶有关节肌肉出血。实验室检查示PT延长、APTT延长,能被贮存正常血浆或血清纠正,不被吸附血浆纠正,TT正常,FⅩ∶C活性降低,蛇毒时间延长是与FⅦ缺乏症鉴别的主要方法。治疗以补充FⅩ为主,可用血浆、PCC或FⅩ的浓缩制剂。
八、遗传性凝血因子Ⅻ缺乏症
本症又称Hageman因子缺乏,临床罕见,属于常染色体隐性遗传,有近亲婚配史,男女均可患病。绝大多数本病患者无出血倾向,甚至手术或外伤后也无异常出血或出血甚微。部分病例伴有心肌梗死及血栓性静脉炎。实验室检查重型患者凝血时间及部分凝血活酶时间延长。FⅫ的定量降低有助于诊断。本病无须治疗,如有出血症状或需手术,可予以替代治疗。
九、遗传性凝血因子
 缺乏症
缺乏症
本症极罕见,为常染色体隐性遗传,男性略多于女性,患者父母往往为近亲婚配。F
 缺乏使血凝块中的纤维蛋白不稳定,易于被纤溶酶降解。纯合子患者血浆中F
缺乏使血凝块中的纤维蛋白不稳定,易于被纤溶酶降解。纯合子患者血浆中F
 低于正常人的1%时则发生出血。延迟性出血为特征性表现,新生儿脐带残端出血及伤口愈合不良多见,严重缺乏者颅内出血较其他凝血因子缺乏症为多。实验室检查F
低于正常人的1%时则发生出血。延迟性出血为特征性表现,新生儿脐带残端出血及伤口愈合不良多见,严重缺乏者颅内出血较其他凝血因子缺乏症为多。实验室检查F
 缺乏时,纤维蛋白凝块极易溶于5mol/L尿素溶液或1%单氯醋酸溶液中,此法可作为本病的筛选试验。F
缺乏时,纤维蛋白凝块极易溶于5mol/L尿素溶液或1%单氯醋酸溶液中,此法可作为本病的筛选试验。F
 含量测定可确诊。治疗较为容易,输少量血浆、血浆冷沉淀物或F
含量测定可确诊。治疗较为容易,输少量血浆、血浆冷沉淀物或F
 浓缩剂就可达到止血效果。
浓缩剂就可达到止血效果。
主要参考文献
1.张之南,郝玉书,赵永强,等.血液病学.第2版.北京:人民卫生出版社,2011.
2.Kanshansky K,Lichtman MA,Coller BS,et al.Williams Hematology.8th ed.New York:McGraw-Hill,Inc,2010.
第十二节
获得性凝血功能障碍
王志梅
一、获得性FⅡ、Ⅶ、Ⅸ、Ⅹ缺乏症(acquired factorsⅡ、Ⅶ、Ⅸ、Ⅹdeficiency)
FⅡ、Ⅶ、Ⅸ、Ⅹ在肝脏合成后,需进一步在肝细胞羧化酶的作用下对其谷氨酸残基进行羧化,方具有凝血功能。维生素K是肝细胞羧化酶的辅因子,故FⅡ、FⅦ、Ⅸ、Ⅹ又称维生素K依赖因子。
【病因】
获得性FⅡ、Ⅶ、Ⅸ、Ⅹ缺乏症的主要病因有:
(一)合成减少
严重肝病引起多种凝血因子合成障碍,其中以FⅡ、Ⅶ、Ⅸ、Ⅹ缺乏最常见。缺乏的程度与肝病的严重程度相一致。
(二)维生素K吸收不良
见于各种原因的胆道阻塞,或术后引流和胆瘘,导致肠道胆汁缺乏,影响维生素K的吸收。长期口服抗生素,肠道细菌群受抑制,以致细菌合成的维生素K不足;小肠吸收不良综合征;严格限制脂肪类食物摄入导致维生素K缺乏症,进而使依赖维生素K凝血因子的谷氨酸残基羧化水平低,影响凝血功能。
(三)维生素K拮抗药的应用
口服抗凝剂如双香豆素和茚满二酮类可阻断维生素K还原,因而抑制维生素K依赖凝血因子的合成。此外敌鼠(diphacinone)是抗凝血杀鼠药,抗凝血作用通过干扰肝脏对维生素K的利用实现。
(四)弥散性血管内凝血
因多种凝血因子大量消耗而严重缺乏。
(五)新生儿出血症
即新生儿因维生素K缺乏所致的出血。
(六)单一的依赖维生素K凝血因子缺乏
①获得性FⅨ缺乏症:肾病综合征患者可从尿中丢失维生素K依赖凝血因子,尤以FⅨ丢失最多,导致FⅨ缺乏;戈谢病患者FⅨ半衰期缩短,使其血浆浓度降低。②FⅩ缺乏症:全身性淀粉样变及急性白血病患者可出现FⅩ的单独缺乏,可能与淀粉样物质吸附或灭活FⅩ有关。应用广谱抗生素除影响维生素K依赖因子的合成外,也可引起单一因子Ⅱ、Ⅶ、Ⅸ、Ⅹ缺乏。
【临床表现】
出血症状轻重不一,与原发病的性质及引起凝血因子缺乏的程度有关。一般表现为皮肤、黏膜自发性出血,也可有血尿、胃肠道出血、月经过多或手术、外伤后出血增多,但未见到深部血肿和关节腔出血。除出血表现外尚有原发病的临床表现。新生儿出血大多发生于出生后2~3天,表现为脐带残端及胃肠道出血,轻症患者4~5天自愈,重者也可发生颅内出血而死亡。
【实验室检查】
PT及APTT延长,而TT正常。FⅡ、Ⅶ、Ⅸ、Ⅹ含量测定最具有价值。注射维生素K 1 5~10mg后24~48小时测定PT,有助于鉴别肝病及维生素K缺乏症。后者凝血酶原时间有明显改善,而前者改善不明显或无改善。
【治疗】
原则上以去除病因和治疗原发疾病为主。
1.对维生素K缺乏症,静脉或肌内注射维生素K 1 即可纠正。长期吸收不良应每周肌内注射维生素K 1 10mg。必要时可输凝血酶原复合物浓缩剂,以补充凝血因子的不足。
2.新生儿出血症并发出血时,可肌内或静脉注射维生素K 1 0.5~10mg,每天1次,连续3~4天。对出血严重的病例,应立即输新鲜血浆或凝血酶原复合物10U/kg,每4~6小时输注1次。
3.双香豆素类药物过量引起出血时,应立即停用抗凝剂,静脉或肌内注射维生素K 1 10~15mg/d,至出血控制为止。严重出血者应输新鲜血浆和凝血酶原复合物,以迅速止血。
4.对严重肝病引起的出血,治疗参照肝脏疾病凝血障碍的有关内容。
二、获得性F
 缺乏症
缺乏症
获得性F
 缺乏症(acquired factor
缺乏症(acquired factor
 eficiency)
eficiency)
较先天性常见,发生机制可为合成障碍、消耗过多或血浆中有抑制物存在。约30%的肝病患者F
 活性降低,淋巴瘤、白血病、尿毒症、系统性红斑狼疮及骨髓瘤等也可引起F
活性降低,淋巴瘤、白血病、尿毒症、系统性红斑狼疮及骨髓瘤等也可引起F
 减少。遗传性FⅧ缺乏症输注血浆后、结核病患者应用异烟肼治疗,以及以往健康者均可产生F
减少。遗传性FⅧ缺乏症输注血浆后、结核病患者应用异烟肼治疗,以及以往健康者均可产生F
 抑制物。临床可表现为轻度自发性出血,但在外伤或手术后出血倾向可能较为显著。实验室检查与先天性F
抑制物。临床可表现为轻度自发性出血,但在外伤或手术后出血倾向可能较为显著。实验室检查与先天性F
 缺乏症相同。治疗主要针对原发病,对急性出血者宜静脉输新鲜血浆,也可用近期库存血治疗,抗体滴度高者予以血浆置换联合免疫抑制治疗。
缺乏症相同。治疗主要针对原发病,对急性出血者宜静脉输新鲜血浆,也可用近期库存血治疗,抗体滴度高者予以血浆置换联合免疫抑制治疗。
三、获得性纤维蛋白原缺乏症
【分类】
获得性纤维蛋白原缺乏症(acquired fibrinogen deficiency)较遗传性纤维蛋白原缺乏症明显多见。
按发病原理可分为三类:①纤维蛋白原合成不足,如严重肝病、晚期癌肿患者;②纤维蛋白原消耗过多,如弥散性血管内凝血、血栓性血小板减少性紫癜、巨大海绵状血管瘤等;③纤维蛋白原溶解,导致纤溶系统过度活化或抑制物减少的任何原因,都会产生纤维蛋白原过度溶解。原发性纤维蛋白原溶解症(primary fibrinogenolysis),简称原发性纤溶,有先天性和获得性两类。获得性原发性纤溶可见于许多疾病,如心、肺、前列腺、子宫及胰腺等器官含有丰富的组织型纤溶酶原激活剂,上述部位的手术、外伤或晚期癌肿均可引起t-PA直接入血,激活纤溶系统;腺癌(尤其是前列腺癌、胰腺癌)、急性早幼粒细胞白血病等肿瘤细胞可释放纤溶酶原活化物,其中以u-PA为常见,也可促发原发性纤溶。所谓继发性纤溶是指体内先有血栓形成或弥散性血管内凝血(DIC)引起微血栓形成,纤溶被激活,因此继发性纤溶主要见于DIC。
【临床表现】
除原发病的临床表现外,可有严重的出血症状。患者原先可无出血,而于分娩或手术中大量出血或渗血不止,血液可以完全不凝固或仅凝成很细小疏松的血块。可同时伴有皮肤、黏膜的大片瘀斑或体腔出血。
【实验室检查】
(一)纤维蛋白原缺乏
纤维蛋白原含量降低,CT、PT、APTT、TT均可延长甚至不凝,正常血浆或正常纤维蛋白原均可以纠正。
(二)纤维蛋白(原)溶解
不论原发或继发性纤溶均可见血纤维蛋白原显著降低,血及尿中纤维蛋白(原)降解产物(FDP)含量增高。纤维蛋白肽Bβ 1-42 和Bβ 15-42 是纤维蛋白原和纤维蛋白最早期的降解物,也可升高;D-二聚体是交联纤维蛋白的降解物,仅见于继发性纤溶,可作为区别原发性与继发性纤溶的指标。纤溶酶-纤溶酶抑制物复合物(PAP)也可增高。
【治疗】
积极治疗原发病,纤维蛋白原严重减少需输注纤维蛋白制剂;存在纤维蛋白溶解亢进者采用抗纤溶药物:①6-氨基己酸:首次静脉注射4~6g,以后每1~2小时静脉滴注1g;②氨甲环酸:250~500mg/d静脉注射或滴注;③对羧基苄胺:400~800mg/d注射或滴注。体外循环中发生纤溶亢进出血者,可选用抑肽酶。
四、肝脏疾病凝血障碍
异常出血是严重肝脏疾病的常见症状之一,出血的原因往往是多因素的,血小板减少、血小板功能障碍、凝血因子合成减少、纤溶活性增强、异常纤维蛋白原血症、维生素K缺乏、DIC、感染等均与出血有关,出血的严重程度与肝脏实质细胞功能损伤的程度呈正相关。
【分类】
(一)凝血因子合成减少或同时合成异常凝血因子
除FⅢ、FⅧ、FⅣ(Ca
2+
)、F
 α链以外,其余所有凝血因子均由肝实质细胞合成,肝细胞功能严重受损可以导致上述凝血因子合成缺少。凝血因子减少的程度与低蛋白血症的严重程度呈正相关。部分患者因维生素K缺乏,进而导致维生素K依赖性凝血因子功能障碍。纤维蛋白原通常在肝功能严重损害或合并DIC时才明显减少。重症肝病时伴随蛋白质合成功能紊乱,肝脏可能合成异常纤维蛋白原,影响正常的止凝血功能。
α链以外,其余所有凝血因子均由肝实质细胞合成,肝细胞功能严重受损可以导致上述凝血因子合成缺少。凝血因子减少的程度与低蛋白血症的严重程度呈正相关。部分患者因维生素K缺乏,进而导致维生素K依赖性凝血因子功能障碍。纤维蛋白原通常在肝功能严重损害或合并DIC时才明显减少。重症肝病时伴随蛋白质合成功能紊乱,肝脏可能合成异常纤维蛋白原,影响正常的止凝血功能。
(二)抗凝血因子合成减少
抗凝血酶(AT)、蛋白C(PC)系统是人体抗凝系统最重要的组成部分,AT主要由肝细胞合成;PC、蛋白S(PS)在肝脏合成且依赖维生素K,故严重肝病时AT、PC、PS含量减少且可同时伴有后两者结构及功能异常。另外,纤溶酶原、α 2 -纤溶酶抑制剂(α 2 -PI)在严重肝病时也会合成减少。
(三)纤溶活性增强
有研究显示肝硬化患者组织型纤溶酶原活化剂(t-PA)含量5倍于正常对照,其原因为伴发的内皮细胞功能损伤,内皮细胞来源的t-PA和尿激酶型纤溶酶原活化剂(u-PA)活性增强,肝脏清除和灭活t-PA、u-PA的能力减低,导致纤溶活性增强,其结果使凝血因子的进一步消耗性减少,产生的FDP具有抗凝血作用,加重了出血的危险或程度。
(四)引起凝血异常的其他原因
肝病时肥大细胞产生的肝素增多,肝脏因肝素酶产生减少对肝素的灭活减少,结果血浆中肝素和类肝素物质浓度增高,加重出血;内毒素血症在肝病中有较高的发生率,内毒素通过损伤肝细胞、血管内皮细胞、直接激活血小板、趋化白细胞等多种途径加重止凝血功能紊乱。
(五)并发弥散性血管内凝血机制
在致病因素如病毒、免疫复合物、内毒素等作用下,内皮细胞受损、内皮下胶原暴露启动内源性凝血途径并激活血小板,肝细胞受损释放大量组织凝血活酶样物质启动外源性凝血途径,肝功能障碍时抗凝血因子合成减少,肝脏灭活活化凝血因子的能力下降,上述种种原因都可导致DIC的发生;内毒素血症及假性神经介质的存在在DIC的发生中起到参与及推动的作用。
(六)血小板减少
是肝病出血的原因之一,多数患者是因为脾功能亢进所致,部分患者血小板减少有免疫因素参与,部分有血小板功能异常。
综上所述,肝脏不仅在凝血因子、生理性抗凝物质及纤溶成分的合成中有着重要作用,也通过灭活激活的凝血因子、t-PA等调节凝血及纤溶,虽然因抗凝血因子生成减少、激活的凝血因子灭活减少及α 2 -PI合成减少使理论上有血栓形成的可能,但最终凝血因子减少、纤溶活性增强占主导地位,临床上表现为出血。
【实验室检查】
(一)血小板计数
肝脏疾病时因为血小板因素引起出血的机会不多,除非为严重的血小板减少,而在这种情况下往往伴有其他原因或并发症。
(二)凝血时间检查
PT、APTT为常规检查项目;肝脏疾病时除FⅢ、Ⅳ、Ⅷ以外,所有凝血因子的合成均减少,故PT、APTT均可延长,但PT延长的程度往往大于APTT。
(三)FDP和D-二聚体
在肝脏疾病时FDP和D-二聚体常常是升高的,反映的是机体处于一种纤维蛋白溶解增强状态,并不一定是DIC的标志。因此肝脏合并DIC时FDP的诊断水平要高于其他因素所致的DIC,>60mg/L方有意义。
(四)抗凝因子检查
AT、PC、PS、纤溶酶原、α 2 -PI因肝脏合成减少,血液中含量下降;t-PA、纤溶酶血浆水平升高,纤溶酶原活化剂抑制剂(PAI)含量下降,上述检查结果改变越明显提示肝损越重。
(五)循环抗凝物质检查
TT延长且能被甲苯胺蓝纠正或血浆肝素浓度增高,提示体内存在肝素样抗凝物质。
(六)肝脏疾病合并DIC
由于肝脏在多个环节参与凝血与抗凝过程,因此肝脏疾病合并DIC的诊断标准不同于其他疾病,不同的实验室检查结果如下:①血小板<50×10 9 /L,或有2项以上血小板活化产物增高;②血浆纤维蛋白原<1.0g/L;③血浆FⅧ∶C水平<50%;④PT延长>5秒;⑤3P试验阳性,或FDP>60mg/L,或D-二聚体水平明显增高。
【治疗】
1.补充凝血因子 输注新鲜冰冻血浆,可补充所有凝血因子及生理性抗凝血成分。输注PCC可有效地提高凝血因子浓度、改善出血,但不能纠正FⅤ减少,且其中含有微量激活的凝血因子,尤其是因子Ⅸa和Ⅹa,而肝病患者的抗凝能力差,因此PCC输注有导致血栓栓塞的可能,多用于有严重出血的肝病患者。输注纤维蛋白原或冷沉淀,可维持纤维蛋白原含量在1.5g/L以上。
2.对血小板严重减少者可输注单采血小板。
3.其他治疗 补充维生素K 1 可使部分患者的凝血异常得到改善;硫酸鱼精蛋白用于治疗肝素或类肝素过多效果好,通常1mg鱼精蛋白中和1mg肝素;原发性纤溶患者可予以抗纤溶治疗。
肝病时应慎用影响血小板功能的药物;慎用可能损伤胃黏膜的药物。
五、获得性循环抗凝物质增多
【病因与发病机制】
广义的获得性循环抗凝物质包括医源性抗凝物质、凝血因子抑制物及血液中存在的肝素样抗凝物质三种情况。
(一)医源性抗凝物质
主要为肝素及双香豆素类抗凝药两种。肝素作为抗凝血酶Ⅲ的辅因子,能加速抗凝血酶Ⅲ对凝血酶、FⅩa、FⅪa等的灭活作用。若循环中肝素增多,则凝血时间及部分凝血活酶时间延长。双香豆素类抗凝药对肝内羧基化酶有抑制作用,使维生素K依赖性凝血因子谷氨酸残基的羧基化水平降低、凝血功能低下,凝血酶原时间延长。
(二)凝血因子抑制物
指患者体内存在的凝血因子抑制物,可以中和某一正常凝血因子的活性进而影响血液凝固。这些凝血因子抑制物通常为IgG,产生机制有两种:一是遗传性凝血因子缺乏症患者多次输注血制品后产生针对“外来抗原”的免疫反应;二是免疫异常患者产生针对凝血因子的自身抗体。临床上以FⅧ抑制物最多见,针对其他凝血因子的抑制物较少。
1.FⅧ抑制物
根据病因可分为血友病AFⅧ抑制物、非血友病AFⅧ抑制物两种。前者多见于部分重型血友病A患者输注含FⅧ血制品后,患者的年龄、制剂纯度、特异性人类白细胞抗原(HLA)与易感性有关,发生率占血友病患者的5%~21%;非血友病AFⅧ抑制物又称获得性血友病,可伴随自身免疫性疾病如系统性红斑狼疮、类风湿关节炎、溃疡性结肠炎以及支气管哮喘、药物反应、恶性肿瘤、妊娠等,FⅧ抑制物可见于部分老年人,既往无基础疾病。因子Ⅷ抑制物绝大多数属IgG,少数为IgG和IgM混合存在。
2.其他凝血因子抑制物
1%~2%的血友病B患者输FⅨ制剂可产生FⅨ抑制物,偶见于自身免疫性疾病患者,正常人罕有发生。这种抑制物大多数为IgG,FⅨ可以迅速中和这种抗体。FⅤ的抑制物多见于老年人,结核感染、输血、手术、某些药物的应用与发病有关,而发生于遗传性FⅤ缺乏者很少,多数患者的抗体为多克隆IgG。FⅡ、Ⅶ、Ⅺ、Ⅻ及纤维蛋白原抑制物均少见。vWF抑制物的发生率不低,发生在3型vWD的临床价值不大;获得性vWD综合征可导致vWF减少而引起出血表现。
(三)血液中的肝素样抗凝物质
见于严重肝病时肝素降解减少、恶性肿瘤细胞分泌、流行性出血热及SLE等疾病时内皮细胞过量释放类肝素样物质。
【临床表现】
通常表现为遗传性凝血因子缺乏,患者出血加重,替代治疗不易奏效,非出血性疾病患者除原发病的表现外出现出血症状,出血的轻重因抗凝物质对凝血因子的灭活程度而定,抗FⅧ抗体严重者可使FⅧ活性下降至1%以下,出现典型的血友病出血症状,可因严重的出血死亡。出现FⅤ及FⅨ抑制物者的临床出血症状多数较轻,但外伤或手术后可能出血较重。
【实验室检查】
患者CT、APTT及复钙时间延长,复钙交叉血浆凝固时间测定有助于抗凝物质存在与凝血因子缺乏症的区别,后者血浆中加入少量正常人血浆即能纠正,而有抗凝物质者则不能纠正。检测抗凝物质滴度及做抗体中和试验有确诊价值。肝素样抗凝物可做TT及甲苯胺蓝纠正试验证实。
【治疗】
积极治疗原发病,部分患者可自发缓解。存在FⅧ、FⅨ抑制物患者的治疗,参阅血友病治疗的相关部分。其他凝血因子抑制物引起出血时可输新鲜血浆和近期库存血浆中和抗体止血,糖皮质激素及免疫抑制剂对系统性红斑狼疮引起的抗体有时可能有效。鱼精蛋白治疗肝素样抗凝物质常有明显疗效。肝素过量也可用鱼精蛋白治疗,静脉注射1mg可以中和1mg肝素。双香豆素类药物过量的治疗见“获得性FⅡ、Ⅶ、Ⅸ、Ⅹ缺乏症”。
主要参考文献
1.王振义,李家增,阮长耿,等.血栓与止血基础理论与临床.第3版.上海:上海科学技术出版社,2004.
2.张之南,郝玉书,赵永强,等.血液病学.第2版.北京:人民卫生出版社,2011.
3.Kanshansky K,Lichtman MA,Coller BS,et al.Williams Hematology.8th ed.New York:McGraw-Hill,Inc,2010.
第十三节
弥散性血管内凝血
刘立根
弥散性血管内凝血(disseminated intravascular coagulation,DIC)是一种在严重原发病基础之上,以血管内凝血为特征,机体广泛的微血栓形成,伴随继发性纤维蛋白溶解亢进的获得性全身性血栓-出血综合征。DIC并非一独立疾病,而是继发于严重疾病的病理过程。由于血管内皮细胞损伤,血小板活化,凝血反应启动,从而导致弥散性血管内凝血,特别是毛细血管内的微血栓形成。在这一过程中,血小板和凝血因子因大量消耗而减少,继发性纤溶亢进又导致凝血因子大量降解,产生具有抗凝血活性的纤维蛋白(原)降解产物,从而引起多脏器栓塞和功能衰竭,广泛严重的全身出血,顽固性休克,微血管病性溶血性贫血。大多数DIC起病急骤,病情复杂,发展迅猛,诊断困难,预后凶险,如不及时识别处理,常危及患者生命。
【病因】
(一)病因
1.严重感染性疾病
包括细菌、病毒、真菌、螺旋体及原虫感染等。革兰阴性杆菌最为常见,如脑膜炎双球菌、大肠埃希菌、铜绿假单胞菌等;某些革兰阳性菌如金黄色葡萄球菌;偶有结核菌的报道。病毒感染包括出血热病毒、肝炎病毒以及风疹和麻疹病毒等。立克次体、疟原虫、钩端螺旋体和组织胞浆菌等病原体感染也是DIC的病因。
2.恶性肿瘤
多种造血系统肿瘤如急慢性白血病、淋巴瘤,其中发病率最高的是急性早幼粒细胞白血病;其他实体瘤以肺癌、胰腺癌、前列腺癌、肝癌多见,且广泛转移者更易诱发DIC。
3.病理产科
为急性DIC常见病因,包括妊娠高血压综合征、羊水栓塞、前置胎盘、胎盘早剥、死胎滞留及感染性流产等。
4.外科大手术及严重创伤
特别是涉及富含组织因子的器官如肺、前列腺、胰腺、肾上腺、颅脑手术,联合器官移植及严重创伤如多发性骨折、挤压伤综合征、严重烧伤等,均可诱发DIC。
5.内科与儿科疾病
各种原因所致休克;恶性高血压;严重缺氧;重症肝病及急性胰腺炎;急性肾小管坏死及肾病综合征;溶血性贫血;糖尿病酮症酸中毒和系统性红斑狼疮;动脉瘤和Kasabach-Merritt综合征(巨大海绵状血管瘤)、主动脉和其他大血管缩窄、Takayasu动脉炎;大的人造动脉移植物、发绀型先天性心脏病等。
6.医源性因素
包括药物、手术等医疗操作;肿瘤放射治疗和化学治疗;溶血性输血反应;细菌污染性输入;严重输液反应;中药或大量非等渗性液体输入所致溶血等。
(二)诱因
1.休克
休克为DIC之表现,亦是DIC的发病诱因,主要原因包括:①血流动力学的紊乱,血流缓慢;②多种生物介质活化血小板,激活凝血过程;③组织细胞的缺氧坏死,引起组织因子释放;④合并代谢性酸中毒;⑤血管通透性增加,血浆外渗,引起血液浓缩及黏滞度增高。
2.酸中毒
败血症合并酸中毒,DIC发生率增加3~4倍。酸中毒诱发DIC机制:①血液凝固性升高;②血小板聚集性增强;③酸性代谢产物对内皮细胞损伤。
3.单核-巨噬系统功能受抑
严重肝病、脾切除术后、肾上腺皮质激素大量应用可封闭单核-巨噬细胞功能,降低其清除已激活凝血因子的能力。
4.缺氧
组织坏死细胞溶解,内皮细胞损伤,组织因子释放。
5.妊娠
妊娠期多种凝血因子水平增高如高纤维蛋白原血症、血小板活性增强、纤溶活性减低、血流动力学异常均影响DIC发生。
【发病机制】
(一)ISTH/SCC(国际血栓与止血学会)微血管体系损伤的作用
1.血管壁损伤
在各种病因如缺血、缺氧、内毒素、抗原抗体复合物、酸中毒等作用下,血管内皮细胞发生两种变化:轻度损伤(亦称内皮激活)主要涉及其功能变化,包括:①vWF合成释放增加;②PAF释放;③合成FⅤ、HMWK,表达TF;④合成分泌PAI。重度损伤表明血管壁结构破坏,包括:①血小板黏附于胶原;②伴随血小板黏附聚集出现血小板释放反应;③TF合成和活性增加;④抗凝蛋白(AT-Ⅲ、PC、PS)含量及活性下降。无论内皮激活还是血管壁结构缺损均导致血浆内皮素升高。
2.血小板活化
包括血小板聚集直接形成血小板血栓;刺激花生四烯酸代谢与TXA 2 生成;活化的血小板释放PF 3 促进凝血;血小板释放 ADP和5-HT加速诱导血小板聚集及缩血管作用。
3.凝血途径的激活
凝血途径的激活是DIC发病机制中最重要的一环。组织损伤、内毒素血症、感染等可使组织因子及其类似物释放入血而启动外源性凝血过程,抑制TF/Ⅶa可完全阻断内毒素诱导的凝血过程,以免疫方式耗竭血浆组织因子途径抑制物(TFPI)可使动物对凝血酶诱导的DIC敏感化,使用TF单抗可抑制DIC发生发展;血管内皮受损,因子Ⅻ和内皮下胶原组织发生接触激活而启动内源性凝血过程,细菌内毒素、血浆中游离饱和脂肪酸、抗原抗体复合物等可直接激活因子Ⅻ。
4.抗凝系统受损
AT是最主要的凝血抑制物,其血浆水平下降,一方面由于激活的中性粒细胞释放弹性蛋白酶的水解作用,另一方面则由于AT的生成受到干扰;PC系统的破坏导致以产生活化蛋白C(APC)来维持血液循环中抗凝系统稳定的能力下降;DIC患者存在获得性TFPI的不足或功能缺陷。
5.纤维蛋白溶解系统功能紊乱
DIC早期凝血系统被激活,而由于血管内皮细胞持续高表达PAI-1,同时缺氧使t-PA合成减少,PAI-1释放增加导致纤溶系统极度受抑;晚期DIC可产生继发性纤溶亢进。
(二)凝血-炎症-免疫损伤的作用
尽管DIC的发病机制非常复杂,但是始终是以凝血酶生成为中心关键环节。鉴于组织因子(TF)在凝血反应启动中的关键作用,其对DIC发病中的主导作用也逐步被认识;同时,炎性细胞因子也在DIC发病的多个环节中发挥作用;免疫性血栓形成(immunothrombosis)失衡通过TF启动了凝血途径,参与了DIC的发病。
1.TF在DIC发病中的主导作用
现代的凝血理论确立了TF启动作用的重要性。外科大手术、创伤、产科意外导致TF直接释放入血;细菌感染、内毒素血症、抗原抗体复合物、炎症因子可以激活机体单核-巨噬细胞和血管内皮细胞以跨膜蛋白形式表TF,从而启动外源性凝血系统;内皮细胞损伤后,内皮下胶原的暴露,由Ⅻ因子启动内源性凝血过程;抗凝血酶系统、蛋白C系统、组织因子通路抑制剂系统的缺陷,三者共同作用导致凝血功能失衡,凝血酶过度产生,导致广泛的微血栓形成。与此同时,凝血过程消耗大量的凝血因子和血小板,并激活纤维蛋白溶解系统,进一步发生消耗性低凝和继发性纤维蛋白溶解亢进从而引起微血栓形成、广泛出血、休克和微循环障碍等一系列临床表现。
2.炎症因子在发病中的作用
体外实验表明多种细胞因子可以调节血管内皮细胞和单核-巨噬细胞的TF表达:TNF、IL-1α、IL-1β、IL-6、IL-8、MCP-1可以上调 TF 表达;TGF-β、IL-4、IL-10、IL-13可以抑制多种因素介导的TF表达增加。IL-6是人体最具代表性的细胞炎症因子,动物实验证实,IL-6可使内皮细胞TF表达增加10倍,IL-10可抑制这一效应;TNF能刺激内皮细胞生成及分泌TF,下调凝血酶调节蛋白(TM),抑制蛋白C激活,抑制纤溶系统。
细胞因子对蛋白C和蛋白S的作用可解释DIC病理过程中抗凝系统的缺陷,TNF和IL-1可以降低培养的内皮细胞TM的活性及基因的表达;TNF也可降低内皮细胞的内皮细胞蛋白C受体(EPCR)的表达及信号传导;IL-1β可以促进EPCR由内皮细胞上脱落、抑制蛋白C的活化;注射重组的TNF可以降低肝肾及睾丸组织蛋白C的mRNA表达,IL-1亦有类似作用,表明TNF和IL-1可以降低多种组织蛋白C的表达而影响凝血过程。
细胞因子对纤溶系统的影响在DIC实验动物模型中得到解释,纤维蛋白溶解起初被激活,但随后被抑制,主要是由于内皮细胞释放纤溶酶原激活剂抑制物-1(PAI-1)增加所致,这种改变是通过TNF和IL-1介导。凝血酶激活的纤溶抑制物(TAFI),如同PAI-1一样,可能在抑制纤维蛋白溶解和加速微血管血栓形成中发挥作用。在DIC患者中发现存在与凝血酶生成相应的极低TAFI水平,特别是感染相关病例,因此,TAFI可能与PAI-1一起涉入了微血管血栓形成诱导的缺血导致多器官功能障碍。
3.免疫性血栓形成的失衡
生物在遭受外伤后,血液凝固防止血液的丢失的同时,也使得外来病原体局限化,以此发挥防御功能,此为免疫性血栓形成。这种作用失去平衡则可导致病理性血栓形成。生物病原体相关分子模式(PAMPs)和损害相关分子模式(DAMPs)与DIC的发生和进展存在密切关系。
【病理生理改变】
(一)微血栓形成
是DIC最本质的病理变化。血栓成分早期为血小板血栓,随后大量的纤维蛋白沉积形成血小板/纤维蛋白血栓,此后红细胞包绕形成混合血栓。微血栓的发生部位广泛,以肺、心、脑、肾最为多见,并引起相应的功能改变。DIC微血栓形成的主要原因包括:①血小板活化、聚集形成血小板血栓;②酰键式纤维蛋白聚体形成。③内毒素、缺氧、酸中毒致内皮细胞脱落,形成小块堵塞血管;④可溶性纤维蛋白单体复合物(SFMC)在PF 4 及粒细胞释放的某些蛋白作用下沉积于微循环。
(二)凝血障碍
是DIC最常见的病理变化。可分为三个阶段:①初发性高凝期:为DIC早期改变,以血小板活化、黏附聚集并释放大量血小板因子、凝血酶及纤维蛋白大量形成为特征。②消耗性低凝期:以血小板、纤维蛋白原、凝血酶原及其他因子因广泛微血栓形成而大量消耗,从而以血栓形成过程减弱为特征。③继发性纤溶亢进期,以凝血过程中因子Ⅻa激活激肽释放酶,进而激活纤溶酶原,微血栓刺激血管内皮细胞释放t-PA使纤溶系统激活而实现,临床上以广泛再发性出血倾向为特征。
(三)微循环衰竭
微循环衰竭与DIC互为诱因,是DIC最常见的后果。DIC休克机制如下:①因子Ⅻa激活激肽和补体系统。激肽、缓激肽及由此诱生的EDRF、PGI 2 及某些补体碎片(C3a、C5a等)使微动脉及毛细血管前括约肌舒张,外周阻力显著下降,导致低血压。②PAF产生,导致血小板活化及释放反应,参与休克的发生;③凝血纤溶产物:大量纤维蛋白肽A(FPA)及肽B(FPB)可引起微静脉及小静脉收缩;FDP引起血管舒张,毛细血管通透性升高,血浆外渗,导致休克的发生。
(四)微血管病性溶血
①缺氧与酸中毒使红细胞可塑变形能力降低;②微血栓形成,可塑性降低的红细胞在通过纤维蛋白网时受到挤压而破碎;③败血症DIC时,内毒素与纤溶碎片D激活补体系统,引起白细胞的趋化反应,产生大量自由基,使红细胞代谢及结构发生改变,导致溶血。
【临床表现】
DIC除原发病表现外,常见有四大临床表现,即出血、休克、栓塞和溶血。
(一)出血
出血是DIC最引人注意的表现,发生率达80%~90%。DIC出血常有以下特点:①早期表现穿刺部位瘀斑或出血不止或试管血不凝固;②最常见的为皮肤自发性出血,表现为瘀点瘀斑,甚至大片广泛紫癜伴中心皮肤黏膜栓塞性坏死;③不能用原发病解释的多部位(一般至少2个部位)、多脏器的自发性出血;④严重者可致颅内出血且常为DIC致死病因;⑤单纯补充凝血因子不仅不能纠正出血,反而加重病情,而适当采用抗凝辅以补充凝血因子和血小板治疗,可取得较好效果。
(二)休克
休克与低血压是DIC又一主要表现,也是DIC诊断依据之一,常发生于革兰阴性菌败血症患者。DIC时休克本身无特殊性,但由于继发于严重基础疾病之上,易被基础疾病的临床征象所掩盖而不易识别。DIC休克一般有以下特点:①起病突然,早期找不到明确病因;②常伴有全身多发性出血倾向,但与出血症状不相称;③早期出现重要脏器的功能障碍;④休克较顽固,常规抗休克治疗效果不佳。
(三)微血栓形成
多发性微血栓形成必然是DIC最早期的表现之一,但可能较隐匿,不易识别。皮肤黏膜微血栓表现为血栓性坏死,主要特点为全身出血性皮肤瘀斑进展为界限清晰的紫黑色皮肤坏死;肺微血栓常导致急性呼吸窘迫综合征,表现为不明原因的呼吸快、低氧血症;肾微血栓引起急性肾衰竭,表现为少尿、无尿;心脏微血栓轻者表现为不明原因的心搏加快,重者导致心功能不全及急性心肌梗死;脑组织受累可表现为神志模糊、嗜睡与昏迷等。广泛的微血栓形成也是引起多脏器功能衰竭(multiple organ function failure,MOFF)的重要因素。
(四)微血管病性溶血
患者可出现不明原因的与出血程度不成比例的贫血症状,可并发寒战、高热、黄疸、血红蛋白尿等,外周血出现较多的红细胞碎片(>2%)或(和)畸形红细胞。微血管病性溶血也可在急性肾衰竭、血栓性血小板减少性紫癜、肿瘤广泛性转移、恶性高血压等疾病中出现,所以在考虑溶血与DIC的关系时,应加以鉴别。
【实验室检查】
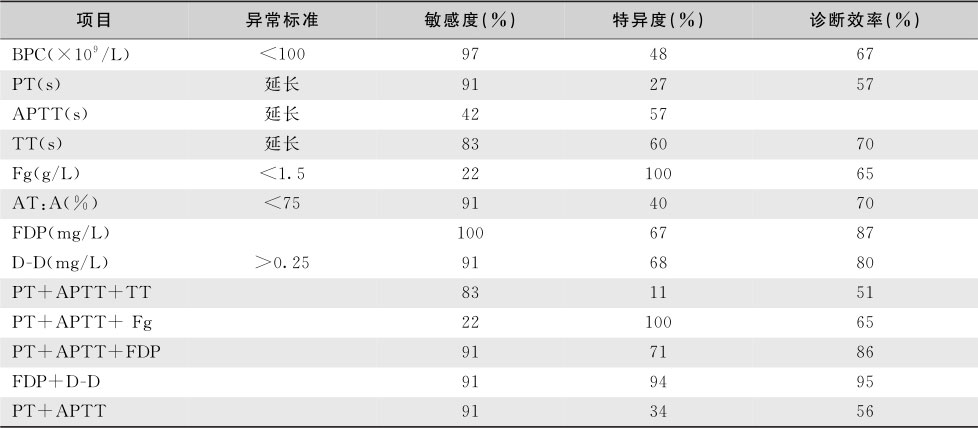
DIC实验室检查主要针对其病理过程中的血管壁(血管内皮细胞为主)、血小板数量及质量、凝血和抗凝系统及纤溶的变化进行检测,这对DIC的诊治有至关重要的意义。由于DIC表现缺乏特异性,常与基础疾病的表现重叠,多数DIC的判断需有实验指标的支持;DIC的多种检查项目不具备高度特异性,检查结果需密切结合临床综合分析,动态观察十分重要;鉴于DIC的危重性,对实验室检查要求简单实用,先易后难,超过90%的患者可通过血小板计数、活化的部分凝血活酶时间(APTT)和凝血酶原时间(PT)、纤维蛋白原(FIB)定量、3P试验及D-二聚体确诊。
(一)血管内皮细胞的检验
1.血浆内皮素-1(ET-1)测定
ET-1是血管内皮细胞损伤的分子标志物之一,正常参考值<5ng/L,其参与DIC的发病和发展过程,并可能与预后有关。
2.血管性血友病抗原(vWF∶Ag)测定
采用免疫火箭电泳法,参考值(94.1±32.5)%,因检测耗时不适于急诊应用。
3.血浆凝血酶调节蛋白(TM)活性测定
采用发色底物法,参考值(100±13)%,敏感性高,可用于前DIC诊断,DIC好转时TM迅速下降,有助于疗效判断。
(二)血小板检查
1.血小板计数
血小板数目减少是DIC中最常见且重要的实验室异常,若血小板计数正常,诊断难以成立。动态观察血小板进行性减少更有价值。
2.血小板活化的分子标志物改变
β-TG、PF 4 存在于血小板颗粒中,是血小板特有的蛋白质,可作为血小板体内活化的指标,急性DIC时增高尤为显著,对慢性或代偿性DIC诊断意义更大;PF 4 可与血浆游离肝素结合,DIC时血栓形成导致血浆肝素样物质减少,因此PF 4 升高可作为广泛血小板聚集活化的指标;P选择素是血小板α颗粒膜外显糖蛋白,其水平的变化可反映血小板活化的程度;TXB 2 是花生四烯酸代谢启动的分子标志物,在急性DIC的早、中期其水平显著升高,后期由于血小板数量减少,逐渐下降至正常,在慢性或代偿性DIC,TXB 2 也有较大的诊断意义。
(三)血浆凝血因子的检查
1.APTT和PT
分别反映内、外源性凝血过程的改变。DIC时由于凝血因子的广泛消耗,APTT和PT可有不同程度的延长,两者同时延长诊断意义更大。
2.纤维蛋白原(FIB)
DIC时纤维蛋白原减少甚为多见,严重者可呈乏纤维蛋白原血症状态,但是由于纤维蛋白原在体内代谢快、代偿能力强且为急性时相反应蛋白,因此在慢性、亚急性DIC,甚至急性DIC早期纤维蛋白原可正常,甚至升高,所以观察纤维蛋白原水平动态变化更有意义。
3.组织因子
TF是凝血反应(特别是病理性)的始动因子,对评估前DIC、早期DIC尤为重要。
4.因子Ⅴ、Ⅶ
因子Ⅴ是组成凝血活酶所必需的消耗性因子,因子Ⅶ是外源性凝血途径中必需的非消耗性因子,两者均产生于肝脏。DIC时因子Ⅴ呈消耗性减少,因子Ⅶ理论上并不减少,以此与肝病两者合成障碍性减少相鉴别。
5.因子Ⅷ
DIC时Ⅷ∶C减低发生率为60%~80%,早期Ⅷ∶C可有暂时性升高,中后期因子Ⅷ虽有消耗,但Ⅷ∶C仍在正常低限;在慢性DIC,因生成加速也罕见Ⅷ∶C下降。
6.因子Ⅹ
组成凝血活酶的重要成分,DIC时呈消耗性减少,其异常敏感性明显高于PT、APTT和纤维蛋白原等指标。
7.分子标志物
血浆凝血酶原片段1+2(F 1+2 )是凝血酶原转变为凝血酶过程中最早释放出来的片段,它直接反映凝血酶生成的总量;FPA反映凝血酶水解纤维蛋白原的活性。两者均有助于前DIC、早期DIC的诊断。
(四)抗凝物质检测
1.血浆抗凝血酶(AT)活性测定
DIC时AT与凝血酶结合而呈消耗性减少,敏感性达90%,对前DIC及早期DIC诊断意义更大。但AT由肝脏生成,故对重症肝病性DIC诊断价值有限。
2.血浆蛋白C(PC)、蛋白S(PS)测定
PC和PS在发病过程中明显下降,其主要原因在于消耗性减少及肝功能受损的生成障碍,但由于其依赖于维生素K合成,因此在维生素K缺乏及肝功能不良患者,PC和PS不宜作为DIC实验诊断指标。
3.血浆组织因子途径抑制物(TFPI)测定
TFPI抑制TF/Ⅶa的活性,DIC时存在TFPI的调控不足。
4.血浆凝血酶-抗凝血酶复合物(TAT)测定
AT-Ⅲ与产生的凝血酶迅速结合形成TAT,从而时使凝血过程减弱,TAT反映凝血酶与抗凝血酶结合形成复合物的量,间接提示凝血酶的生成,是前DIC及早期DIC敏感指标之一。
(五)纤溶活性检查
1.血浆鱼精蛋白副凝固试验(3P试验)是临床上常用的可溶性纤维蛋白单体复合物(SFMC)定性试验,它反映凝血和纤溶两个病理过程的存在。DIC血浆中出现的SFMC主要是纤维蛋白单体与FDP中的碎片X所组成的复合物,鱼精蛋白可使此复合物解离,纤维蛋白单体聚合形成纤维蛋白丝胶状物,此称为副凝固现象。本试验阳性,主要表明血液中有SFMC存在;而血清鱼精蛋白副凝固试验阳性,才表明有FDP增多。碎片X是一种分子量较大的早期降解产物,在DIC早期,纤溶系统尚未启动,血浆内无足够的FDP和SFMC产生;而晚期,由于继发性纤溶亢进,体内无过量的纤维蛋白单体存在,碎片X极少,而分子量较小的晚期降解产物Y、D、E增多,此类小碎片不能与纤维蛋白单体形成SFMC,因此在这两种情况下,3P试验可呈阴性结果。此外血液中医源性肝素增多,可干扰鱼精蛋白的作用,导致3P试验假阴性。在手术、创伤的情况下,血液中凝血酶及纤溶酶水平增加,可导致3P试验假阳性。
2.优球蛋白溶解时间(ELT)血浆优球蛋白组分中含有Fg、PLG和PA,但不含纤溶酶原抑制物。在pH为4.5时可使优球蛋白沉淀,将此沉淀溶解于缓冲液中,再加Ca 2+ 或凝血酶使其凝固;在37℃条件下观察凝块完全溶解所需要的时间。参考值为120分钟以上。DIC时,如纤溶亢进,则ELT缩短;反之,则提示纤溶活性降低。
3P试验和ELT检查历史悠久,但两者共同的缺点在于敏感性低,比如,在典型的DIC患者中,ELT的阳性率仅在20%~30%,在三级医院中已经逐步被其他检查所替代,在基层医院中仍有其价值。
3.FDP反映血液中纤维蛋白(原)在纤溶酶作用下生成X(x)、Y(y)、D(d)、E(e)碎片的含量,DIC时阳性率85%~100%,诊断有效率75%,血清FDP>20mg/L,对继发性纤溶有诊断价值。
4.D-二聚体 D-二聚体增高表明体内有纤维蛋白的形成及纤溶的发生,其敏感性及特异性均较高,是目前诊断DIC有价值的指标之一。
5.血浆纤溶酶原(PLG)活性 血浆纤溶酶原活性降低,表明其被消耗而提示纤溶活性增强。
6.血浆纤溶酶与抗纤溶酶复合物(PAP)在DIC的早期PAP可正常或轻度下降,而在继发性纤溶亢进期,PAP明显上升。
7.血浆纤维蛋白肽Bβ 1-42 和Bβ 15-42 测定 前者为纤维蛋白原的降解产物,后者是纤维蛋白的降解产物,两者升高表明纤溶酶的激活,是DIC的敏感指标之一。
8.SFMC定量 反映凝血和纤溶两个病理过程的存在,对DIC的早期诊断极有价值,与3P试验相比,本试验更直接、敏感、特异。
(六)中性粒细胞细胞外捕获网(neutrophil extracellular traps,NETs)和DNA结合蛋白
DIC时,病原体或炎症通过PAMPs和(或)DAMPs活化中性粒细胞释放NETs,血浆NETs增高。组蛋白H2A、H2B、H3和H4是主要的染色质组成成分,DIC时血浆水平增高。高迁移率族蛋白-1(High mobility group box 1 protein,HMGB1)是一种典型的DAMPs分子,DIC时显著增高,与患者DIC积分和预后相关。
(七)评价
DIC实验室检查项目繁多,临床上除考虑检查的敏感性和特异性外,很多项目耗时费力,因此对其实用性提出更高的要求。
国外DIC研究机构通过荟萃分析5个独立的临床研究得出结论,诊断项目出现异常的概率由高至低分别为血小板减少、纤维蛋白降解产物增加、PT延长、APTT延长、纤维蛋白原降低。
血小板减少或进行性下降是诊断DIC敏感非特异的指标,血小板计数低和凝血酶生成密切相关,因血小板消耗是由凝血酶诱导的血小板聚集所致。
FDP和D-二聚体是反映继发性纤维蛋白溶解亢进的指标,前者是纤维蛋白原和交联纤维蛋白单体的降解产物,而后者仅为交联纤维蛋白单体的降解产物,相比较而言,D-二聚体对诊断DIC更有特异性。但由于在外伤、近期手术、血栓性疾病时,两者均会升高;且FDP可经肝脏代谢与肾脏分泌,肝肾功能异常可干扰FDP水平,因此这两项指标均不宜作为单独诊断DIC的标准,必须结合其他检查。SFMC产生于血管内,外界影响小,其诊断DIC敏感性几乎达100%,但特异性低,ISTH在诊断评分系统中建议使用SFMC代替D-二聚体可以增加诊断的特异性。
APTT和PT在50%以上的患者疾病的某一阶段存在着延长,换言之,近半数DIC患者PT和APTT正常或缩短,这是由于活化的凝血因子(如凝血酶或因子Ⅹa)所致,因此PT和APTT正常不能排除凝血系统的激活,必须动态监测。
纤维蛋白原属于急性时相反应蛋白,尽管在 DIC时持续消耗,但其血浆水平仍可在正常范围,临床上,典型DIC病例中,纤维蛋白原降低的敏感性不足30%,对DIC诊断帮助不大。有关细胞游离DNA及其结合蛋白测定尚处于研究阶段,对于肿瘤和创伤相关的DIC诊断意义较大,具有良好应用前景 (表20-7-8)。
表20-7-8 DIC常用实验室指标及其评价
【分型与分期】
1.根据患者病理过程,可以分为血栓形成为主型和纤溶过程为主型(表20-7-9)。
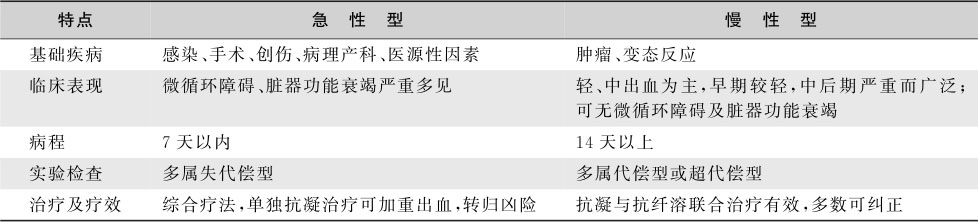
2.根据临床经过分为急性型和慢性型(表20-7-10)。
表20-7-9 血栓形成为主型与纤溶过程为主型的DIC主要特点

表20-7-10 急性型与慢性型DIC的不同特点
3.根据患者内环境调节功能紊乱的情况,DIC可分为代偿性DIC和失代偿性DIC。ISTH/SSC将DIC分为两型,显性DIC与非显性DIC,前者包含了既往分类命名的急性和失代偿型DIC,后者包含了慢性和代偿性DIC及DIC前期。
4.临床分期 DIC在临床上分4期,即临床前期、早期、中期及后期。①临床前期亦称前DIC(pre-DIC),指在基础病因下,体内凝血纤溶系统发生一系列变化,但尚未出现典型DIC症状及体征,或尚未达到DIC确诊标准的一种亚临床状态。此期特点为血液呈高凝状态,血小板的活化、凝血过程已经开始但尚无广泛的微血栓的形成,纤溶过程尚未或刚刚启动,血小板、凝血因子的消耗均不明显。此时如能及时识别,对DIC的防治有重要意义。②早期DIC,属于病理过程中的初发性高凝期。③中期DIC,属于病理过程中的消耗性低凝期。④后期DIC,属于病理过程中的继发性纤溶亢进期。
【诊断】
国际、国内关于DIC的诊断标准众多,在此主要介绍ISTH/SSC的诊断积分系统和国内标准。
(一)ISTH/SSC推荐的诊断积分系统
1.显性DIC诊断积分系统
(1)风险评估:
患者是否存在与典型DIC发病相关的基础疾病,包括:
1)严重感染(任何微生物)。
2)创伤(多发性创伤、神经损伤、脂肪栓塞)。
3)器官损伤(重症胰腺炎)。
4)恶性肿瘤(实体瘤、骨髓/淋巴组织恶性增殖性疾病)。
5)产科意外(羊水栓塞、胎盘早剥)。
6)血管异常(大血管动脉瘤、Kasabach-Merritt综合征)。
7)严重肝衰竭。
8)严重中毒或免疫反应(毒蛇咬伤、输血反应、移植排斥)。
如果存在,则进入下一步评估。
(2)进行实验室检查,包括:
1)血小板计数。
2)凝血酶原时间。
3)纤维蛋白原。
4)可溶性纤维蛋白单体,或纤维蛋白降解产物等。
(3)评分:
1)血小板计数(10 9 /L):>100=0,50~100=1,<50=2。
2)纤维蛋白相关标志物升高:不升高=0,轻度升高=1,显著升高=2。
3)凝血酶原时间延长:<3s=0,3~6s=1,>6s=2。
4)纤维蛋白原水平:>1g/L=0,<1g/L=1。
(4)结果判定:
将以上各分数相加后,如果≥5分,符合显性DIC,每天重复积分;如果<5分,提示(非肯定)非显性DIC,随后1~2天重复积分。
2.非显性DIC诊断积分系统
(1)风险评估(同上)。
(2)实验室检查
1)血小板计数。
2)凝血酶原时间。
3)纤维蛋白降解产物。
4)蛋白C活性。
5)抗凝血酶活性等。
(3)评分
1)血小板计数(10 9 /L):增加=-1,减少=1。
2)凝血酶原时间延长:不延长=-1,延长=1。
3)D-二聚体:不升高=-1,升高=1。
4)蛋白C活性(%):正常=-1,降低=1。
5)抗凝血酶活性(%):正常=-1,降低=1。
(4)结果判定:将以上各分数相加后,如果≥5分,符合非显性DIC,随后1~2天重复积分。
该积分系统以基础疾病为前提,检测项目简单易行,对诊断DIC敏感性为91%,特异性为97%,评分≥5分时诊断DIC阳性预测值为96%,评分<5分时排除DIC的阴性预测值为96%,适用范围广,目前其诊断价值已得到广泛的认同。
(二)中国DIC诊断标准修订方案(第八届全国血栓与止血学术会议,2001年,中国武汉)
1.一般诊断标准
(1)存在易于引起DIC的基础疾病,如感染、恶性肿瘤、病理产科、大型手术及创伤等。
(2)有下列两项以上临床表现:①严重、多发性出血倾向;②不易以原发病解释的微循环衰竭或休克;③广泛性皮肤、黏膜栓塞、灶性缺血性坏死、脱落及溃疡形成,或不明原因的肺、肾、脑等脏器功能衰竭;④抗凝治疗有效。
(3)实验室检查符合下列标准:在上述指标存在的基础上,同时有以下三项以上异常①血小板<100×10 9 /L或进行性下降;②纤维蛋白原<1.5g/L或呈进行性下降或>4.0g/L;③3P试验阳性或FDP>20mg/L或D-二聚体水平升高(阳性);④凝血酶原时间缩短或延长3秒以上或呈动态性变化,或者活化的部分凝血活酶时间延长10秒以上;⑤疑难或其他特殊患者,可考虑行抗凝血酶、因子Ⅷ∶C及凝血、纤溶、血小板活化分子标记物测定:血浆纤溶酶原(PLG)<300mg/L,抗凝血酶AT活性<60%(不适用于肝病)或PC活性降低,血浆ET-1含量>80ng/L或TM增高,血浆凝血酶片段F 1+2 、凝血酶-抗凝血酶复合物(TAT)或纤维蛋白肽A(FPA)含量增高,血浆可溶性纤维蛋白单体复合物(SFMC)含量升高,血浆纤溶酶-纤溶酶抑制物复合物(PIC)水平升高,血浆组织因子(TF)含量增高(阳性)或组织因子通路抑制物(TFPI)水平下降。
2.肝病合并DIC的实验室诊断标准
(1)血小板<50×10 9 /L或有两项以上血小板活化产物(β-TG、PF 4 、TXB 2 、P-选择素)。
(2)纤维蛋白原<1.0g/L。
(3)血浆凝血因子Ⅷ∶C活性<50%(必备)。
(4)凝血酶原时间延长5秒以上或者呈动态性改变。
(5)3P试验阳性或FDP>60mg/L或D-二聚体水平升高(阳性)。
3.白血病并发DIC的实验室诊断标准
(1)血小板<50×10 9 /L或血小板活化、代谢产物水平增高。
(2)纤维蛋白原<1.8g/L。
(3)凝血酶原时间延长5秒以上或者呈动态性改变。
(4)3P试验阳性或FDP>40mg/L或D-二聚体水平显著升高(阳性)。
4.基层医院DIC的实验室诊断参考标准(具备下列三项以上异常):
(1)血小板<100×10 9 /L或呈进行性下降。
(2)纤维蛋白原<1.5g/L或进行性下降。
(3)3P试验阳性或FDP>20mg/L。
(4)凝血酶原时间缩短或延长3秒以上或者呈动态性改变。
(5)外周血破碎红细胞比例>10%。
(6)血沉<10mm/h。
总之,DIC诊断中以下四点值得注意:①高度重视是否存在引起DIC的基础疾病。②DIC的症状或体征往往存在于基础疾病之中,有些难以识别;DIC出血往往发生于多个部位,很少单一部位。③DIC的实验室检查缺乏特异性,不能单凭某一项或者某几项试验来确诊DIC。建议用BPC、Fg、PT、FDP、D-D和AT作为诊断DIC的基本试验,以分子标志物检测为补充。④动态观察实验室检查指标对诊断和疗效判断有重要意义。
【鉴别诊断】
(一)重症肝病
重症肝病的出血机制较为复杂,涉及内皮损伤、血小板减少、激活、凝血因子减少、纤溶亢进多种因素,临床上与重症肝病诱发DIC较难鉴别。肝病合并DIC的诊断较其他疾病引起的DIC有更加严格的要求,其中引人注意的是因子Ⅷ的变化。目前认为因子Ⅷ可能由肝脏间质组织等单核巨噬细胞系统合成,在肝病时尽管大多数凝血因子合成减少,活性下降,但是缘于库普弗细胞功能亢进,因子Ⅷ活性增强;内皮损伤导致vWF水平升高。肝病合并DIC时,由于凝血因子的消耗,因子Ⅷ和vWF水平下降。所以,因子Ⅷ活性高低是单纯肝病性出血和肝病合并DIC鉴别诊断的要点之一。Ⅷ∶C<50%以上或动态下降是肝病合并DIC诊断必不可缺少的条件。
(二)原发性纤维蛋白溶解亢进
本病罕见,在出血倾向、纤维蛋白原水平低下及纤溶亢进方面与DIC十分相似,但本病不涉及血小板的活化和下降,无凝血反应的启动和内皮细胞损伤,D-二聚体作为交联纤维蛋白之降解产物,理论上只见于DIC,有鉴别诊断意义。
(三)血栓性血小板减少性紫癜
该病以血浆vWF裂解酶ADMATS13先天或获得性缺乏为基本病因,以血小板血栓形成为主要病理变化,临床上以血小板减少性出血、微血管病性溶血、神经精神症状、发热和肾功能损害为特征,表现与DIC有较多相似之处。但本病休克和呼吸衰竭少见,微血管病性溶血重,无凝血及纤溶系统的激活,血浆置换可奏效,检测血浆ADMATS13活性及其抑制物有助于鉴别诊断。
(四)抗磷脂综合征(APS)
临床上具有反复发作的血栓形成、习惯性流产、血小板减少三大表现,伴随神经症状、肺动脉高压、皮肤表现(网状青斑、下肢溃疡、皮肤坏死、肢端坏疽)等,实验室检查可见抗磷脂抗体阳性、抗心磷脂抗体阳性、狼疮样抗凝物质阳性、β 2 糖蛋白1抗体(抗β 2 -GP1)阳性。
【治疗】
DIC的主要治疗措施包括:去除病因和诱因;根据临床分期,干预DIC病理生理过程,阻断血管内凝血过程,恢复正常的血小板和血浆凝血因子水平,抗纤溶治疗;对症支持治疗。
(一)治疗原发病、消除诱因
原发病的治疗是终止DIC病理过程的关键。积极控制感染,抗生素应足量早期联合应用,选择敏感杀菌药物。对于革兰阴性菌感染,应考虑到抗生素诱导的内毒素释放效应,应尽可能使用低诱导内毒素释放的抗生素。积极抢救休克,改善微循环,主要措施有补充血浆容量,输血浆、白蛋白、葡聚糖等,合理应用血管活性药物。纠正酸碱失衡、电解质紊乱及缺氧,改善心肌代谢、增强心肌收缩力。
(二)根据临床分期进行分层治疗
1.DIC早期(弥散性微血栓形成期)——抗凝治疗
抗凝治疗是阻断DIC病理过程的最重要措施之一,目的在于抑制广泛性微血栓形成,防止血小板和凝血因子的进一步消耗,为重建凝血-抗凝平衡创造条件。
(1)普通肝素:
肝素治疗DIC的机制主要包括:①抑制凝血因子Ⅻa、Ⅺa、Ⅸa活性;②抑制因子Ⅹa对凝血酶原的激活,在肝素辅因子(HC-Ⅱ)存在条件下肝素结合AT后可与凝血酶形成复合物,降低凝血酶活性;③肝素与血管内膜结合使内皮细胞释放t-PA,促进纤溶活性;④通过抗血小板聚集作用,使凝血活性受抑;⑤肝素诱导TFPI活性,抵抗TF作用。
肝素的剂量选择:既往强调“足量”,近年来随着对肝素作用认识的深入、制剂的改进和综合治疗措施的应用,已趋向于小剂量用药。多数学者认为,①首剂50~100U/kg,一般5000U,静脉滴注,每6~8小时半量重复,皮下注射,以APTT调整用量,根据病情连续使用3~5天,适用于急性DIC患者;②前述剂量半量使用,或每日总量200U/kg,分3~4次给药,皮下注射,每疗程8天,适用于慢性DIC患者;③每日总量10~15U/(kg·h),持续静脉滴注可逆转DIC的病理过程而无严重出血危险,无须血液学监测,适用于急性DIC患者;④每日总量50U/kg小剂量应用,分3~4次给药,皮下注射,连续5~8天,适用于DIC预防。
肝素治疗时血液学监护:①CT(试管法):CT正常在8~12分,肝素的有效治疗应控制CT在正常高限的2倍左右,即25分钟;超过30分,意味肝素过量;低于15分,则肝素用量不足。②APTT:控制APTT较正常延长1.5~2.5倍意味用量适宜。
肝素的剂量调整:①根据DIC的临床类型和病期,急性型、重症DIC早期,肝素用量适当增加;②酸中毒时,肝素灭活快,用量宜偏大;③肝素在肝脏代谢,50%由肾排除,肝肾功能障碍时,用量宜小;④血小板重度减少,凝血因子明显低下时,应减少肝素用量;⑤血浆AT减少时,肝素用量增加,但应提高AT水平。
肝素治疗有效指标及停药指征如下。提示肝素治疗有效:①出血停止或逐步减轻。②休克改善或纠正。③尿量增加。④PT比治疗前缩短5秒以上,纤维蛋白原及血小板计数不再进一步下降或有不同程度的回升。⑤其他凝血现象检查逐步改善。停药指征:①诱发DIC的原发病已控制或缓解。②临床上病情改善明显,如停止出血、休克纠正、有关脏器恢复正常。③PT缩短到接近正常,纤维蛋白原升到1.0~1.5g/L以上,血小板数量逐渐回升或至少不再下降。④APTT超过肝素治疗前2.5倍以上;或PT超过30秒;凝血酶时间超过50秒;APTT延长接近100秒。⑤出现肝素过量的表现。
肝素无效的原因:①病因未去除;②血小板因素:血小板大量破坏,PF 4 大量释放于血液循环,拮抗肝素的作用。③AT减少:因肝素的抗凝作用是通过AT发挥的,故此造成肝素作用减弱。
(2)低分子量肝素(LMWH):
DIC凝血的启动几乎均首先形成Ⅹa,再形成凝血酶。一般认为抗凝治疗中,抗Ⅹa活性与其抗凝能力密切相关,而抗凝血酶活性则与用药后出血并发症有关。鉴于LMWH抗Ⅹa作用远大于抗凝血酶活性(4∶1),而普通肝素为1∶1,因此LMWH抗DIC疗效优于普通肝素。
LMWH用法:①预防:每日总量50~100U/kg,分2次皮下注射,疗程5~10天或更长。②治疗:每日总量200U/kg,分2次皮下注射,疗程5~8天。为预防治疗相关性出血,可以行抗Ⅹa活性试验检测,使其维持在0.4~0.7U/ml的最佳治疗剂量;也可用APTT监测,标准同普通肝素。
2.DIC 中期(消耗性低凝血期)——抗凝联合替代治疗
此期微血栓仍在形成,抗凝治疗必不可少,但因凝血因子进行性消耗,所以应充分抗凝基础上,进行血小板和凝血因子的替代治疗,适当输注新鲜全血、新鲜血浆、纤维蛋白原、血小板悬液、凝血酶原复合物浓缩剂。
新鲜血浆所含凝血因子和新鲜全血相似,并可减少输入液体总量,有助于纠正休克、改善微循环。
纤维蛋白原适用于急性DIC出现低纤维蛋白原血症或出血倾向严重者,首剂2~4g静脉输注,以后根据血浆纤维蛋白原水平而补充,使血浆纤维蛋白原达到1.0g/L以上为宜。由于纤维蛋白原血浆半衰期达96~144小时,所以不需每日使用,建议根据病情每周使用2~3次为宜。
血小板悬液:当血小板≤20×10 9 /L或<50×10 9 /L伴活动性出血时应输注单采血小板。
凝血酶原复合物浓缩剂(PCC):剂量为20~40U/kg,每次以5%葡萄糖液50ml稀释,要求在30分钟内静脉滴注完毕,每日1~2次。PCC缺少因子Ⅴ,而且有可能加重凝血功能紊乱,发生血栓形成,因此应谨慎使用,密切观察,同时应注意到输注PCC 6个小时内应避免使用抗纤溶药物。
3.DIC晚期(继发性纤溶亢进期)
此期微血栓形成已经基本停止,继发性纤溶为主要矛盾,若临床确认纤溶亢进是出血的首要原因,则可适量应用抗纤溶药物。鉴于抗纤溶制剂已在临床上广泛使用,因此必须强调,对于有出血倾向而没有排除DIC,或怀疑为DIC所致的患者,不宜将抗纤维蛋白溶解制剂作为首选的止血药予以使用,以免诱发或加重DIC。少数以原发性或继发性纤溶亢进占优势的疾病,如急性早幼粒细胞白血病合并DIC可考虑使用抗纤溶药物,特别提出的是,急性早幼粒细胞白血病标准诱导分化治疗采用全反式维甲酸可增加血栓形成的风险,因此应特别谨慎。
常用的抗纤溶药物包括:①6-氨基己酸(EACA),常用剂量每次4~10g,以5%葡萄糖或生理盐水100ml稀释,静脉滴注,小剂量每日5g以下,中等剂量每日10g以下,大剂量每日可达20g,本品快速静脉注射可引起血压下降,休克者慎用。②氨甲苯酸(对羧基苄胺,PAMBA),每次200~500mg加入葡萄糖液20ml中,静脉注射,每日1~2次。③氨甲环酸,用量为EACA的1/10,小剂量每日0.5g以下,中等剂量每日1.0g以下,大剂量每日可达2.0g。④抑肽酶:抑肽酶兼有抑制纤溶酶和因子Ⅹ等激活作用,呈纤溶、凝血双相阻断,理论上最适合DIC的治疗,常用剂量每日8~10万U,分2~3次使用,或首剂5万U,随后每小时1万U,缓慢静脉注射。
(三)其他可能有益的治疗手段
1.抗凝血酶
AT是一种重要的凝血抑制物,在临床研究中发现,AT可改善DIC患者实验室参数、减少出血、纠正凝血异常,但不能降低病死率。因此在欧洲和日本对AT使用存在明显分歧:英国指南不推荐使用,而日本专家共识却强烈推荐。
2.组织因子通路抑制剂
TFPⅠ抑制TF的活性,在败血症患者中开展的TFPⅢ期临床试验中显示了预期的治疗效益,但在Ⅲ期临床中,患者存活率未显示有显著改善。
3.活化蛋白C
蛋白C系统在DIC发病中起了重要作用,补充APC可能对DIC治疗有益。在动物实验中,应用APC制剂显示能有效降低病死率和器官功能衰竭。但是在最近的关于APC治疗成人感染性休克的临床研究结果显示,即使在严重的蛋白C缺乏的感染性休克病例中,使用APC也不能降低死亡率,因此应完善有效的治疗方案,改善预后仍任重而道远。
主要参考文献
1.宋善俊,王鸿利,李家增.弥散性血管内凝血.第2版.上海:上海科学技术出版社,2001,355-365.
2.Beutler E,Lichtman MA,Coller BS,et al.Williams hematology.8th ed.Columbus:McGraw-Hill,2010,1677-1697.
3.Wada H,Asakura H,Okamoto K,et al.Expert consensus for the treatment of disseminated intravascular coagulation in Japan.Thrombosis Research,2010,125(1):6-11.
4.Kaneko T,Wada H.Diagnostic criteria and laboratory tests for disseminated intravascular coagulation.J Clin ExpHematop,2011,51(2):67-76.
5.Engelmann B,Massberg S.Thrombosis as an intravascular effector of innate immunity.Nature Rev Immunol,2013,13(1):34-45.
6.Gando S,Otomo Y.Local hemostasis,immunothrombosis,and systemic disseminated intravascular coagulation in trauma and traumatic shock.Critical care,2015,19:72.
第十四节
血栓形成与血栓栓塞性疾病概述
王伟光 程韵枫
血栓是血液成分在血管或心脏内膜表面形成的凝块或沉积物。血栓栓塞性疾病指由于先天遗传性或后天获得性原因,导致患者止血和抗血栓机制失衡,引起血凝块阻塞血管的疾病。高凝状态包括一系列引起病理性血栓形成倾向和血栓形成风险的遗传或获得性病变,亦称为前血栓形成状态。血栓并非一种永久的结构,它经历延伸和滋长、溶解、机化、再通和钙化、栓塞不同的病理过程,该组疾病是当今世界上致病、致畸、致死的主要原因。
【血栓形成机制】
早在1845年德国病理学家Virchow就提出血栓形成机制三要素学说,即血管壁的损伤,血流的紊乱和血液成分的异常。在此基础上,经过160多年的临床和实验研究,并随着近年来分子生物学、免疫学和生物化学的发展,对其发病机制有更丰富的认识。
(一)血管壁的损伤
引起血管损伤的原因包括机械因素(血液流动产生的切变应力、血管内压力及机械性损伤);化学物和代谢产物;感染因素(细菌病毒及内毒素血症的作用)及免疫因素。
1.血管内皮细胞的抗血栓形成作用
(1)生成和释放促进血管松弛的物质:主要包括内皮衍生松弛因子(endothelial-derived relaxing factor,EDRF)和前列环素I 2 (prostacyclin I 2 ,PGI 2 )。EDRF实质是内皮衍生的一氧化氮(NO),它激活血小板鸟苷酸环化酶,使cGMP增加;PGI 2 是内皮细胞磷脂代谢产物,它与血小板膜上相应受体结合,激活腺苷酸环化酶,使cAMP增加。cGMP和cAMP协同可发挥舒张血管及抑制血小板聚集作用。
血管内皮细胞结构和功能完整时,血小板对管壁是排斥而被动进入循环的,前列环素和一氧化氮作为强烈的血管扩张剂在局部起到了对血小板的抑制作用。但在血管损伤处,这些活性物质减少,为血小板的黏附聚集提供了基础。
(2)生成和释放抑制血小板黏附和聚集的物质:除EDRF和PGI 2 外,血管内皮细胞上ADP酶能水解血小板诱聚剂ADP,生成AMP和腺苷,后者具有抑制血小板聚集的作用。
(3)生成抗凝类物质:血管内皮细胞膜表面结合的大量硫酸乙酰肝素;分泌抗凝血酶(antithrombin,AT);合成分泌凝血酶调节蛋白(thrombomodulin,TM);产生组织因子途径抑制物(tissue factor pathway inhibitor,TFPI)及细胞表面分布蛋白C(protein C,PC)受体。上述物质均抑制血栓形成。
(4)释放促进纤溶活性的物质:血管内皮细胞合成和分泌组织型纤溶酶原激活剂(tissue plasminogen activator,t-PA)和尿激酶型纤溶酶原激活剂(urokinase type plasminogen activator,u-PA),完整的血管内皮细胞表面存在t-PA和纤溶酶原受体,两者能促进纤溶活性。
(5)血管内皮细胞可摄取或破坏促进血小板聚集的物质如5-羟色胺。
2.血管壁的促血栓形成作用
(1)内皮细胞产生内皮素-1(endothelin-1,ET-1),是目前所知最强烈的缩血管物质。
(2)血管内皮细胞合成和分泌黏附分子包括von Willebrand因子(vWF),Ⅲ、Ⅳ、Ⅴ型胶原及凝血酶敏感蛋白(thrombospondin,TSP),纤维联接蛋白(fibrinectin,Fn)。vWF是参与血小板与血管基底膜黏附的主要蛋白。
(3)内皮细胞产生血小板活化因子(platelet activating factor,PAF)是迄今所知最强的血小板诱聚剂。
(4)血管内皮细胞表达组织因子(tissue factor,TF)。TF是跨膜糖蛋白,在血管外层的平滑肌细胞、成纤维细胞、星形细胞和树突状细胞可恒定表达,以备止血。而正常情况下,血管内皮细胞、单核细胞、中性粒细胞及巨噬细胞不表达TF活性,在病理因素(缺氧、内毒素、IL-1、TNF等)作用下,可启动血管内皮细胞和单核细胞TF诱导性表达,导致血液凝固。
(5)血管内皮细胞表达Ⅺ/Ⅺa活性,并结合活化的Ⅹ(Ⅹa)加速内皮细胞表面凝血酶原的激活。
(6)血管内皮细胞合成分泌纤溶酶原激活物抑制剂-1(plasminogen activator inhibitor-1,PAI-1)。
血管壁的损伤主要导致其抗栓和促栓机制失衡,如促进血小板的黏附聚集和活化(vWF、Fn、PAF释放增多);血管壁的痉挛(ET-1增多,PGI 2 和EDRF减少);促凝活性增强(TF表达);抗凝活性下降;纤溶机制异常。
(二)血流的紊乱
血液是一种非牛顿流体,意即血流缓慢(低切变应力)导致血黏度升高,而黏度升高加重血流缓慢。凝血因子局部的堆积,单核-巨噬细胞系统清除作用受限,易形成静脉血栓。当血流速很快(高切变应力),在血管分叉处易形成湍流,造成血管壁损伤、内皮下胶原的暴露、红细胞内ADP的释放及血小板的活化,易形成动脉血栓。
(三)血液成分的改变
1.血小板的变化
血小板膜表面糖蛋白(GP)Ⅰb在血小板静息的状态下即可与vWF结合而发生黏附反应,而血小板聚集必须依靠血小板的活化才能实现,GPⅡb-Ⅲa的表达是血小板活化的基础。血小板激活方式有三种:①释放5-HT或ADP。血小板受ADP刺激后,GPⅡb~Ⅲa的表达;接触产物生成活性(CFPA)和胶原诱导的凝血活性(CICA)从血小板膜磷脂成分释放,分别激活因子Ⅻ和因子Ⅺ,参与凝血反应。②依赖于花生四烯酸代谢过程中TXA 2 的产生。TXA 2 激活的血小板磷脂酰丝氨酸由膜内向外翻转,形成PF 3 ,启动凝血过程。③胶原和凝血酶刺激血小板释放PAF,促进纤维蛋白原和GPⅡb~Ⅲa的结合。
活化的血小板胞质中Ca 2+ 浓度升高后,其促凝作用体现在以下几个方面:①磷脂酰丝氨酸通过磷脂翻转酶的作用从膜内侧转移到膜表面,与凝血因子Ⅹ、Ⅸa、Ⅷa、Ⅴ相互作用形成凝血酶原酶,从而启动血小板表面的凝血反应。②GPⅢa上的纤维蛋白原受体在静息血小板上是通过凝血酶原上的RGD与凝血酶原结合,在血小板活化时,结合在GPⅢa上的凝血酶原迅速被纤维蛋白原取代后,快速形成凝血酶。③活化血小板表面存在效应细胞蛋白酶受体-1(effector cell protease receptor-1)能与Ⅹa结合,参与凝血反应。④活化的GPⅠb可激活Ⅺ。
血小板由诱导剂和血流切变应力两个方面引起聚集。诱导剂可分为以下几种:①弱作用:ADP、儿茶酚胺、血管加压素;②中等作用:TXA 2 ;③强作用:凝血酶、胶原、PAF。切变应力引起血小板聚集包括以下两种情况,在低切变应力(18m N/mm 3 )下,GPⅡb~Ⅲa、纤维蛋白原、Ca 2+ 参与聚集过程;在高切变应力(108m N/mm 3 )下,除GPⅡb~Ⅲa、纤维蛋白原、Ca 2+ 外,尚有GPⅠb和vWF参与。
血小板功能亢进是引起血栓形成的常见原因,具体表现在:血小板对诱导剂的敏感性升高;释放反应增强(TXA 2 、PF 4 、β-TG的作用);血小板抗纤溶活性增强(血小板合成释放PAI-1,TGF-β加强内皮细胞合成PAI-1)。凡是血管内皮损伤、血流切变应力改变、某些药物和各种疾病(SLE、TTP、DIC、HUS和冠心病等)都可导致血小板活化而形成血栓。另外血小板数量增多特别是超过800×10 9 /L时有明显的血栓形成倾向。
新近的研究表明,血小板膜糖蛋白基因变异与血栓形成也有一定的关系,GPⅢ a 基因的PL A2 多态性与急性血栓形成之间相关;GPⅠ a 基因的α 2 多态性及GPⅠ bα 的Ko多态性、可变数目串联重复序列(VNTR)、Kozak多态性均与血栓形成有关。
2.凝血因子异常
研究表明,纤维蛋白原增加(肥胖、糖尿病、高脂血症等)、因子Ⅶ活性增高(吸烟、口服避孕药、饮酒)是动脉粥样硬化血栓形成的两大独立危险因素。纤维蛋白原及因子Ⅶ结构异常,手术和创伤使凝血因子Ⅷ、Ⅸ、Ⅹ升高均有利于血栓形成。先天性凝血因子Ⅴ、Ⅷ增高患者常伴有自发性血栓形成倾向。
3.抗凝系统减弱
先天性AT-Ⅲ减少或缺乏、获得性AT-Ⅲ消耗过多、蛋白C和S缺乏症以及肝素辅因子Ⅱ(HC-Ⅱ)缺乏,均有利于血栓形成。
4.纤溶活性过低
先天性因子Ⅻ缺乏症导致患者纤维蛋白溶解系统的内激活途径缺陷;广泛的内皮细胞损伤导致t-PA释放耗竭;大手术和严重创伤时纤溶抑制物(α 2 -巨球蛋白)增多;异常纤维蛋白原对凝血酶反应差,形成纤维蛋白缓慢,但形成的异常纤维蛋白对纤溶酶不敏感等因素均易造成血栓性疾病。
5.白细胞因素
白细胞在正常情况下对血管内皮黏附作用轻微,在病理情况下明显增加。粒细胞和单核细胞表面都有黏附受体(一种糖蛋白复合体CD11/CD18),可增加白细胞对内皮的黏附作用,黏附后又释放一系列血管毒性物质(如中性弹力酶、白三烯、氧化物质、IL-1、TNF、胶原酶等),降低了内皮细胞抗栓功能;病理情况下单核细胞表面可诱导性表达TF,启动凝血反应。急性早幼粒细胞白血病,病态的早幼粒细胞释放促凝物质,诱发DIC。
6.副凝现象
指体内非凝血酶依赖性的类纤维蛋白沉积过程。在体内血流较快部位,虽然有凝血酶的产生及纤维蛋白单体的形成,但由于稀释作用,纤维蛋白稳定因子浓度较低,纤维蛋白单体可与纤维蛋白原或纤维蛋白较大的降解产物形成可溶性纤维蛋白单体复合物。这些复合物可以被单核巨噬细胞系统吞噬,在微循环中沉淀下来。PF 4 、中性粒细胞释放的某些蛋白质均可使纤维蛋白单体从可溶性纤维蛋白单体复合物中解离出来,并通过氢键而聚合沉淀,形成微血栓。
【分类】
血栓的主要构成为血小板、白细胞、红细胞和纤维蛋白。按性质与组成可分六类:血小板血栓、白色血栓、红色血栓、混合性血栓、微血栓、感染性血栓。仅以血小板聚集而成的栓子常发生于微血管;血小板栓子伴有纤维蛋白构成白色血栓,见于动脉粥样硬化斑块;红色血栓多发生于静脉,局部血流缓慢为先决条件;混合血栓最常见,包含所有四种成分,可见于动脉、静脉,或心脏部位;微血栓有紧密的纤维蛋白束组成,主要见于DIC;感染性血栓以内皮损伤为基础,血栓中有白细胞或细菌的聚集。临床上将血栓分为动脉血栓、静脉血栓、动静脉血栓和微血管血栓,一般而言,血小板活化是动脉血栓形成的基础,凝血-抗凝异常/血流缓慢易造成静脉血栓。血栓栓塞性疾病的临床分类见表16-7-11。
【诊断】
血栓形成的过程基本可以分两个阶段,一为血栓形成前状态(高凝状态)和血栓形成初期,二为血栓形成期。后者造成脏器缺血坏死,临床表现突出,诊断较容易;而前者有血栓形成倾向或血栓尚不足以影响脏器血液供应,临床表现轻微,诊断困难。因此,预示血栓形成前的高凝状态实验诊断尤为重要。
(一)实验室检查
血栓性疾病常用实验诊断项目见表16-7-12。
表16-7-11 血栓性疾病的临床分类

表16-7-12 血栓性疾病实验诊断项目

血浆D二聚体作为交联纤维蛋白的特异性降解产物之一,是继发性纤溶特有的代谢产物。总体来说,目前临床上使用的D二聚体检测方法局限于低特异性和低阳性预测值,对可疑的静脉血栓形成有很大的局限性,通常对急性深静脉血栓形成灵敏度较高,ELISA法D二聚体>500μg/L有重要的参考价值;而D二聚体<500μg/L即可排除急性或活动性血栓栓塞的可能。
静脉血栓栓塞大多数以遗传性病因为基础,常于成年早期发病,但是从儿童到老年任何时间内,都可能有血栓形成的迹象出现,尤其以下肢静脉血栓或肺栓塞常见。遗传性病因的患者发生动脉血栓的机会少,但高同型半胱氨酸血症例外。过早发生动脉血栓,特别是不伴有明显的危险因子存在,应全方面检查,包括系统性栓塞的可能来源,以及血管炎、骨髓增殖性肿瘤、抗磷脂抗体综合征的筛选甄别。
(二)器械检查
1.血管造影
包括动静脉造影,能显示血栓的部位,但由于是创伤性检查、部分患者碘过敏、检查本身可损伤血管内皮引起血栓形成,故受到一定限制。
2.超声多普勒检查
包括多普勒频谱和彩色多普勒检查。前者可反映血流持续时间、流速、血流信号的强弱及血流的方向;后者可测定血流的方向、流速、血管管径及有无血流存在。
3.螺旋CT和MRI血管成像
可精确诊断实质脏器中血栓形成的梗死病灶,但对肢体血管血栓形成的诊断不理想。
4.数字减影血管造影术
能克服常规血管造影的缺点。
5.放射性核素检查
通过放射性核素标记纤维蛋白原和血小板来检测。
【血栓栓塞性疾病】
(一)易栓症(thrombophilia)
不同于高凝状态或血栓前状态,指患者存在易发生血栓的遗传性或获得性缺陷。易栓症的病因分类见表16-7-13。遗传性的临床特点是有血栓家族史,无明显诱发因素的多发性、反复的血栓形成,年轻时(<45岁)发病,轻微激惹即可发生血栓,对常规的抗血栓治疗效果不佳;而获得性可见于肝病、肾病综合征及系统性红斑狼疮等。先天遗传性易栓症临床特征见表16-7-14。
表16-7-13 易栓症的分类

表16-7-14 先天遗传性易栓症临床特征

续表
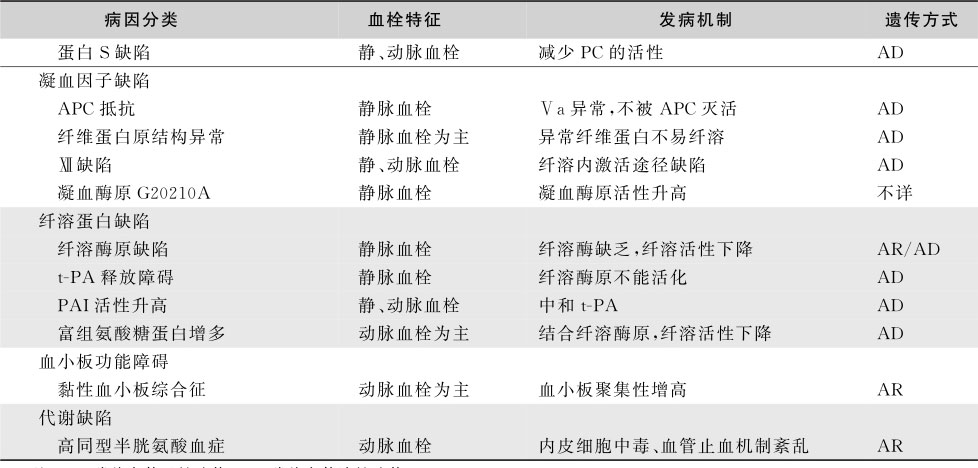
注:AD.常染色体显性遗传;AR.常染色体隐性遗传
对于先天性者,应着重行 AT、PC、PS、活化的蛋白C抵抗(activated protein C resistance,APC-R)、异常纤维蛋白原检测。此外,有些疾病服用某些药物都可能影响AT、PC、PS等活性水平,如血栓性疾病、肾病综合征、DIC、肝病、失蛋白性肠病及应用肝素、雌激素均可导致AT活性降低;活动性血栓病、肝病、DIC、糖尿病及应用华法林、门冬酰胺酶、口服避孕药均可使PC活性降低。
黏性血小板综合征是属于先天性血小板聚集功能增强的易栓症,占不明原因动脉血栓的20%。临床分三型:Ⅰ型:血小板对ADP和肾上腺素均产生高聚集反应;Ⅱ型;血小板仅对肾上腺素产生高聚集反应;Ⅲ型:血小板仅对ADP产生高聚集反应。AT缺陷、PC缺陷、PS缺陷的诊断均有赖于实验室检查。
获得性因素常见的是抗磷脂抗体综合征(antiphospholipid syndrome,APS)是获得性易栓症的最常见原因,由抗磷脂抗体引起的一组相关的临床症候群称为APS。抗磷脂抗体(antiphospholipd antibodies,aPA)在系统性红斑狼疮阳性率约为50%。本症血栓形成的发生率约30%~40%。可能缘于胎盘血管的血栓形成,习惯性流产、胎死宫内、早产和胎儿发育迟缓是APS相关的常见并发症。
(二)恶性肿瘤和血栓
恶性肿瘤患者血栓发生率约为5%~15%,胰腺癌可达50%。主要由于肿瘤细胞促进血小板的激活;单核-巨噬细胞与肿瘤细胞接触后诱导性表达组织因子激活凝血系统;血管内皮细胞的改变;抗凝活性的减弱以及纤溶系统的受抑均为血栓形成的基础。另外,化疗药物、糖皮质激素、感染、手术、创伤性探查也为诱发血栓形成的因素。
(三)系统性红斑狼疮
血栓并发症高达20%,该病除由自身免疫机制损伤血管壁、活化血小板外,aPA在其发病中占主导地位:损伤血管内皮,削弱其抗栓能力;使TM生成减少,影响蛋白C的活化;促进PAF的释放;t-PA生成减少、PAI-1活性升高;抑制PGI 2 合成;使局部产生细胞因子,增加TF的表达从而形成高凝状态。各型冷球蛋白造成高黏滞综合征;循环免疫复合物导致内皮损伤造成局部血栓形成和纤维蛋白沉积。以上种种原因可导致SLE患者发生血栓性静脉炎,皮肤坏死性血管炎,心肌梗死,肺栓塞,缺血性骨坏死等。
(四)心脑疾病和血栓
动脉粥样硬化所致血小板黏附聚集,血管内皮TF的表达、因子Ⅶ及纤维蛋白原增高激活凝血系统,为心脑血管疾病的发病基础。
(五)糖尿病和血栓
糖尿病患者动脉粥样硬化发生率高,冠心病的发生率比正常人群高25倍,国内资料统计占38%,其他可引起视网膜病变、四肢坏疽、脑血管意外等。控制高凝状态是防治血管病变的关键。
(六)肝、肾病和血栓
重症肝病常易引起微血栓,发生DIC。发生率一般为8.8%~16.0%。机制主要是凝血因子激活,抗凝因子合成减少,单核-吞噬系统功能不全所致。肾脏疾病的血栓主要见于肾病综合征,狼疮性肾炎,微小病变和膜性肾炎。
(七)高脂血症和血栓
乳糜微粒(CM)和极低密度脂蛋白(VLDL)中的三酰甘油影响Ⅶ的抗原性及含量;VLDL与t-PA呈负相关,与PAI-1正相关;低密度脂蛋白(LDL)中的胆固醇与血小板的活化、动脉粥样硬化的发生密切相关;LP(a)可直接与纤维蛋白结合,封闭纤溶酶的底物;诸多因素促进血栓的形成。
(八)手术和创伤
不同类型的手术发生率有很大差异,尤其以骨科和神经外科手术的发生率为最高。
(九)药物与血栓
1.肝素
肝素诱导的血小板减少性血栓形成,机制不明,可能与肝素依赖的血小板抗体有关。诊断主要依靠有肝素依赖的血小板减少症,血小板下降30%以上,出现新的血栓并排除其他原因所致。
2.华法林
见下文。
3.抗纤溶药
尤其是在高凝状态、肾功能不全、休克情况下,抗纤溶药应用不当,剂量偏大,疗程过长时造成。
4.避孕药
血栓形成与雌激素有关,通过凝血系统激活、抗凝功能下降、增强血小板的功能、削弱纤溶系统功能导致血栓形成。
5.门冬酰胺酶
使蛋白C、蛋白S、AT减少,凝血-抗凝失衡。
综上,临床上力求详尽的病史、体检,常规的全血细胞计数、血液生化及影像检查,以求早期发现诊断线索。除典型的深静脉血栓和肺栓塞外,游走性浅表血栓性静脉炎(即Trousseau综合征)和非细菌性血栓性心内膜炎提示潜在肿瘤可能;肝静脉、门静脉血栓形成可能反应存在骨髓增殖性肿瘤或阵发性睡眠性血红蛋白尿背景;华法林诱导的皮肤坏死高度提示蛋白C或蛋白S缺乏;反复自发性流产则与APS有关。
【防治】
广义而言,血栓栓塞性疾病的防治主要包括药物治疗和外科手术、介入治疗,前者常用药物主要包括抗血小板、抗凝和溶栓药物;后者包括手术取栓、腔静脉介入下导管接触性溶栓和机械性消栓、临时或永久性腔静脉滤器植入治疗等。本节重点讨论药物治疗。
(一)抗血小板药物(分类见表16-7-15)
表16-7-15 抗血小板药物分类

血小板激活方式主要有三种,ADP受体的兴奋、花生四烯酸代谢、PAF的释放,所造成的结果包括 GPⅡb-Ⅲa的表达、PF 3 的形成和凝血过程的启动。因此封闭ADP受体、阻断花生四烯酸的代谢、拮抗GPⅡb-Ⅲa是抗血小板治疗的重点。
1.阿司匹林
不可逆的抑制环氧化酶1和2,从而阻断TXA 2 的合成。这种作用持续血小板的一生,7~10日。目前多数人认为以小剂量即50~100mg/d已足以抑制血小板聚集,个别患者可应用300mg/d,再大剂量可抑制血管内皮细胞花生四烯酸的代谢,PGI 2 的合成减少,反而有利于血栓形成。
2.双嘧达莫
能抑制ADP诱导的血小板聚集,使血小板cAMP增高;增强动脉壁合成前列环素;促进NO的释放。应用剂量口服25~50mg,每日3次,也可200~400mg/d加生理盐水或5%葡萄糖静脉滴注。
3.ADP受体阻断药
代表药物为氯吡格雷(clopidogrel),氯吡格雷通过选择性抑制ADP与其血小板受体的结合及继发于ADP介导的糖蛋白GPⅡb/Ⅲa复合物的活化而抑制血小板聚集;氯吡格雷还能通过阻断由释放的ADP引起的血小板活化的扩增,抑制其他激动剂诱导的血小板聚集;氯吡格雷通过不可逆地修饰血小板ADP受体发挥作用,使暴露于氯吡格雷的血小板的寿命受到影响。常用剂量为75mg/d,在心绞痛、闭塞性周围动脉疾病及缺血性脑卒中的患者中显示出良好的疗效,该药的不良反应包括出血,胃肠道反应如腹痛、消化不良、胃炎和便秘,皮疹和其他皮肤病,神经系统症状如头痛、眩晕、头昏和感觉异常,肝脏和胆道疾病等。
4.血小板膜GPⅡb/Ⅲa抑制剂
①单克隆抗体:阿昔单抗(abciximab),封闭纤维蛋白原受体,抑制血小板黏附和聚集。在血管成形术前10分钟,静脉滴注250μg/kg,持续1分钟以上,然后以10μg/min的速率维持12小时,然而严重的出血危险性不容忽略。②替罗非班(tirofiban)是一种非肽类的酪氨酸衍生物,为血小板糖蛋白Ⅱb/Ⅲa受体拮抗药,对于血管成形术/动脉内斑块切除术患者开始接受治疗时,应与肝素联用由静脉输注,起始推注剂量为10μg/kg,3分钟内完成,而后以0.15 μg/(kg·min)的速率维持滴注,持续36小时。
(二)抗凝治疗
1.肝素
广泛的抗凝活性是通过与AT的结合,放大AT的作用而实现,对凝血酶的抑制作用强,其适应证主要为预防和治疗各种动静脉血栓栓塞性疾病、DIC、及血栓前期的高凝状态;急性缺血性脑血管综合征、心绞痛及周围血管疾病。一般分三种剂量:①小剂量,24小时成人用量为6000~12 000U,每8~12小时分次应用,不必血液学监测。多用于冠心病心绞痛、高凝状态及预防性给药。②中剂量,24小时总量为20 000U左右,持续静滴或每4~6小时分次给药,适用于血栓栓塞性疾病和DIC。③大剂量,比中剂量增加1倍左右,需监测凝血功能,以APTT延长不超过正常的1.5~2.5倍为宜,此时兼顾了良好的抗凝效果和较少的出血风险,适用于肺栓塞。肝素的疗程一般不超过10天。肝素无效时应考虑以下原因:①病因未去除。②大量血栓已形成如DIC晚期。③血中AT、HC-Ⅱ缺乏或耗竭。④严重酸中毒、缺氧时肝素灭活。⑤大量血小板破坏释出PF 4 和TSP拮抗肝素的作用;肝素主要副作用是出血、血小板减少、过敏反应,长期用药可引起注射部位皮肤坏死和骨质疏松。肝素所致的血小板减少症发生率约5%,轻型为肝素对血小板的直接作用所致,用药2~4天内发生,停用后很快恢复;重型因肝素依赖性抗血小板抗体所致,初用者4~15天内发生,再次用药在2~9天出现,常伴有血栓栓塞和出血,预后不佳。
2.低分子量肝素(LMWH)
指分子量低于12 000的肝素,其特点有:①通过AT抑制Ⅹa的作用较强,所以临床出血倾向较小;②皮下注射吸收完全,生物利用度高,半寿期较长(约4小时),抗血栓能力强;③与PF 4 的亲和力低而不发生中和反应。其适用于不稳定心绞痛、急性脑梗死、DIC、血液透析及防治深部静脉血栓和肺栓塞。
3.磺达肝素
为完全人工合成的戊糖,其设计基础是普通肝素和低分子肝素均包含的天然戊糖结构,通过结构改良,加速Ⅹa复合物的形成,导致Ⅹa的快速抑制。与LMWH相比,磺达肝素半衰期更长(约17小时),生物利用度100%,ACCP2008年静脉血栓防治指南和欧洲心脏病协会2008年急性肺栓塞诊治指南均将该药列为A类推荐药物。预防用药为2.5mg,每日1次;治疗剂量为7.5mg,每日1次。由于其通过肾脏代谢,当内生肌酐清除率<50ml/min时慎用,<30ml/min时禁用。
4.维生素K拮抗剂
主要是香豆素类衍生物和茚二酮衍生物。前者包括苄丙酮香豆素(华法林)、双香豆素、醋硝香豆素等,其中华法林应用最广,该类药物抑制维生素K还原酶的活性而影响维生素K的再利用,在体内影响Ⅱ、Ⅶ、Ⅸ、Ⅹ因子的羧基化而起抗凝作用;后者在结构上类似维生素K,起竞争性拮抗作用。口服抗凝药优点是口服有效,作用时间长,但奏效慢,不易控制,多数用于预防。华法林的首剂剂量为7.5~10.0mg/d,5天后可获得稳定的抗凝作用,其后,以每天2.5~5.0mg/d,使INR维持在2.0~3.0左右。鉴于抗凝因子PC、PS亦为维生素K依赖,且华法林对PC、PS的影响先于凝血因子,所以华法林治疗早期可能有血栓形成倾向,应合用肝素3~5天,以起到抗凝效应。口服抗凝剂的主要副作用是出血,可补充维生素K 1 ,必要时输注凝血酶原复合物或新鲜血浆;偶有皮肤坏死和胆汁滞留性黄疸。使口服抗凝药的敏感性增高的因素:①老年人;②肝功能损害;③发热、甲状腺功能亢进;④服用广谱抗生素、磺胺药、吲哚美辛、保泰松、水杨酸、氯喹、西咪替丁、别嘌醇、奎尼丁、利血平、甲状腺素、胰高糖素、苯磺唑酮等。以下原因可影响该药的疗效:①肠道吸收差;②黏液性水肿;③服用巴比妥类、肾上腺皮质激素、雌激素、灰黄霉素、利福平、苯妥英钠等。口服抗凝剂主要用于高凝状态、心脏换瓣术后、房颤、急性心肌梗死、深静脉血栓形成、大手术或分娩后等情况。在维生素K依赖凝血因子中,Ⅶ半衰期最短,口服抗凝剂最先影响Ⅶ水平,可用反映外源性凝血过程的PT、INR作为观察指标,预防性抗凝要求INR在1.5~2.5,治疗性抗凝INR为2.5~3.5。
5.新型抗凝药物
(1)直接的凝血酶抑制剂:
包括水蛭素类和非水蛭素类,前者无论天然或基因重组,均较肝素类药物有更高的性和有效性,尤其已被FDA批准应用于肾衰竭患者;后者主要为阿加曲班和达比加群酯。
阿加曲班是合成的左旋精氨酸的衍生物,适用于严重肾功能不全和HIT的抗凝治疗;达比加群酯口服吸收,不需要INR监测,适用于HIT患者,2008年达比加群酯被欧盟批准用于静脉血栓的预防和治疗,同年在骨关节大手术的血栓预防中,又获得加拿大的批准。该药80%以原型经肾脏排泄,所以严重肾功能不全患者禁用。
(2)Ⅹa直接抑制剂:
利伐沙班是第一个口服的Ⅹa拮抗剂,对Ⅹa作用具有高度选择性,是其他凝血因子的10000倍,不需要辅因子,任何患者均可口服固定剂量而不需要监测,常规预防剂量为10mg,每日1次,治疗剂量为每日20mg,每日1次,一般维持3~12个月。
表16-7-16 不同抗凝药物的半衰期及清除方式
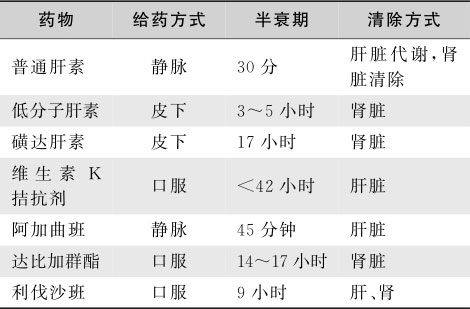
一般而言,重大的静脉血栓和(或)肺栓塞发生后,抗凝治疗疗程至少持续6个月,此后应结合出血并发症的风险加以权衡。先天性易栓症者已有过两次血栓形成事件发生的,应终生接受预防治疗;反复发生血栓形成者,即使未检出病因,也应坚持无限期终生抗凝。
表16-7-17 先天性易栓症的长期处理

(三)溶栓治疗
目前已广泛应用于治疗急性心肌梗死、肺栓塞、深静脉血栓形成、外周动脉血栓形成等。
1.第一代溶栓药物
(1)链激酶(SK):
由β溶血性链球菌产生提取的一种单链蛋白,有间接的纤溶酶原激活作用,形成纤溶酶。由于SK为细菌产物,在治疗后8~9天,产生大量抗SK抗体并在体内维持4~6个月,因此部分患者接受治疗后出现血压下降或皮肤潮红等过敏现象,故主张先注射少量链激酶以观察反应并中和部分抗体,然后用足够剂量静脉滴注。急性心肌梗死以150万U静滴60~90分钟;肺栓塞和新鲜静脉血栓形成,先大剂量25万U静滴20分钟,继以每小时10万U速度静滴24~72小时;急性四肢缺血症状经动脉导管在血栓附近以每小时5000U的速度滴注,直至好转。
(2)尿激酶(UK):
由尿中提取或组织培养人肾细胞制成,直接的纤溶酶原激活剂,体内半寿期约15分钟,无抗原性及过敏反应,一半被肾脏清除,其余由肝脏分解。临床应用参考剂量:急性心肌梗死为200万~300万U;肺栓塞和新鲜深静脉血栓,15万~30万U在12~24小时内滴注;急性四肢端缺血。可用导管介入血栓局部,每小时滴注37万~75万U,后根据纤维蛋白原含量加以调整。
2.第二代溶栓药物
(1)重组组织纤溶酶原激活物(rt-PA):
t-PA于血管内皮细胞中合成,在组织器官中以子宫、卵巢、前列腺、淋巴结、肺脏含量最为丰富。rt-PA来源于黑色素瘤培养液中大肠埃希菌DNA重组,选择性激活血凝块纤溶酶原,半寿期6~8分钟,应持续静滴3~4小时。该药无抗原性及过敏反应,大剂量应用可出现低纤维蛋白原血症,全疗程总量应低于100mg。据报道,rt-PA与肝素或阿司匹林联用疗效优于单用。
(2)重组单链尿激酶(rscu-PA):
有血、尿或条件培养液中提取,半衰期很短,只能静脉滴注,价格昂贵,目前仅小规模临床应用。
(3)乙酰化纤溶酶原-链激酶复合物(APSAC):
是一种经cDNA重组技术制成的链激酶-纤溶酶原复合物的改良型溶栓剂,其本质为选择性长效SK制剂,半衰期长,可一次性静脉推注给药,可提高纤溶效果的选择性,缺点是可引起过敏反应和被抗体中和活性。治疗急性心肌梗死的推荐剂量为30mg,在5分钟内一次推注。
3.第三代溶栓药物
是正在开发中的新型药物,目的是提高选择性溶栓效果和延长天然型溶栓药物的半衰期以减少药物的剂量。包括改造自然型t-PA的分子结构,组建嵌合型(t-PA和scu-PA)溶栓剂,单抗导向溶栓剂,葡激酶的开发等。
四、外科手术、介入治疗
对于股静脉切开手术取栓、腔静脉介入下导管接触性溶栓和机械性消栓,需严格掌握适应证和禁忌证,关于临时或永久性腔静脉滤器植入治疗,更应充分评估远期并发症的风险,采取慎之又慎的态度。
主要参考文献
1.王振义,李家增,阮长耿.血栓与止血基础理论与临床.第3版.上海:上海科学技术出版社,2004.
2.刘泽霖,贺石林,李家增.血栓性疾病的诊断与治疗.北京:人民卫生出版社,2000.
3.王鸿利,王学锋.血栓病临床新技术.北京:人民军医出版社,2003.
4.阮长耿.出血与血栓性疾病的诊断和治疗进展.临床血液学杂志,2010,23(1):1-4.
5.赵永强.易栓症的研究概况.中国实用内科杂志,2007,27(1):49.
6.Goldman L,Schafer AI.Goldman-Cecil Medincine.25th ed.Philadelphia:Elsevier Saunders,2016,1185-1190.

