第二节
人全基因组、全外显子组和全转录组测序的基本原理和基本步骤及其检测报告

一、人全基因组测序
(一)全基因组从头测序(de novo sequencing)
如果某物种的参考基因组(reference genome)的序列未知,对其全基因组进行序列测定,然后运用生物信息学,对所得的大量序列进行拼接、组装,从而获得该物种的全基因组序列图谱的测序方法称为全基因组从头测序。例如,通过2003年完成的人类基因组计划,获得了第一个人的参考基因组。
(二)全基因组重测序(resequencing)
参考基因组序列已知的物种的不同个体进行基因组测序,然后在个体或群体水平上进行差异性分析的测序方法。将某个体测序后获得的大量序列,与该物种的已知参考基因组序列比对,可以确定该个体包含的各种类型的突变,包括单碱基变异、插入缺失、结构变异及拷贝数变化。通过对个体间基因差异的分析对比,并结合不同个体的表型,可做遗传进化分析,确定重要性状以及功能的相关基因。近年来,随着二代测序技术的迅速发展和成熟,测序试剂成本降低至1000美元以下,人全基因组重测序已成为研究人类疾病快速有效的方法之一。可以在全基因组水平上检测与疾病相关基因突变位点及结构变异等信息,开发精准治疗药物。
1.全基因组重测序的两个前提条件
(1)已知该物种的参考基因组序列。
(2)所测定群体的个体之间遗传相似度很高(>99 %)。
2.全基因组重测序的基本步骤
以Illumina的双末端测序方法(pair-end sequencing)为例(图2-5-4 A)。
(1)文库制备(library preparation):
提取基因组DNA,随机打断后,通过连接酶在每个DNA片段的两端加上测序接头,经PCR扩增后,纯化并回收有接头的DNA片段(图2-5-4A-a)。
(2)簇生成(cluster generation )(图2-5-4A-b):
在有序流动槽上进行。步骤如下:
1)文库模板杂交:
文库经碱变性,形成大量单链DNA后,引入流动槽,与流动槽纳米孔表面的接头序列(adapter)杂交,固定在流动槽的表面。
2)桥式扩增:
固定在纳米孔表面的DNA单链的另一端与纳米孔表面的相应接头杂交,形成桥状结构。然后每条单链经DNA聚合酶催化,合成互补链,形成桥式双链。双链再变性成两条单链,进入下一个桥式扩增,重复多个循环后,每条单链扩增至上千条,形成一簇。大量的簇在纳米孔表面的内壁上随机分布,呈超高密度簇。
3)单链化:
簇由DNA双链构成,定向切断接头序列上的切割位点,产生一条单链,游离单链经碱变性后,冲洗去除。
4)末端封闭:
DNA单链3'游离末端加上一个dd NTP,封闭末端,以防测序时的随机延伸。
5)引物结合:
加入测序引物,与每条单链DNA的相应位点结合。
(3)测序(图2-5-4 A-c):
在Illumina测序仪上进行。
1)碱基延伸:将流动槽反应物转移到测序仪上进行碱基延伸。所用d NTP带有四种荧光基团之一和终止基团,因此每个循环只能延伸1个碱基。洗去未结合的荧光标记的核苷酸。
2)荧光信号激发与采集:荧光基团经激光激发后,产生荧光信号,用特殊相机拍摄。
3)末端切除:拍照后,用酶切除末端碱基的荧光基团和终止基团。单链3'末端碱基不再产生荧光,恢复到可延伸的状态。
4)重复上面三个步骤,进行第二个碱基的信号收集,直至完成所有循环。
(4)序列比对(sequence alignment)与数据分析(图2-5-4A-d)
1)荧光信号数据分析:
将每个循环中拍摄的照片按时间顺序排列,然后对每个扩增簇进行坐标定位,根据每个簇的颜色变化,进行碱基识别并读取一条序列。读取序列的长度取决于测序的循环次数,并用分析软件对每个碱基进行质量评估,给予每个碱基一个质量分值(quality score,Q-score)。
2)序列比对与后续分析:
将每个序列和参考基因组序列进行比对,然后进行各种后续分析,包括序列组装和拼接、序列中各种基因突变类型的识别定量等。基因组数据分析的主要参数有:①读取的序列:DNA测序读取的每一条序列,包含A、T、G、C四种碱基。每一个扩增簇产生一个读取的序列,如10 000个扩增簇产生10 000个单向读取序列(single reads),或20 000个双向读取序列(paired-end reads)。②读取序列的总量:样品基因组测序读取的序列总量。③测序覆盖度:测序后,样品中的每个碱基与参考基因组中相对应的碱基进行比对后,得到的每个碱基平均被测的次数。如果某样品的全基因组重测序的测序覆盖度是30,即该基因组中的每个碱基平均被测了30次。④测序深度:测序得到的总碱基数量与基因组碱基总数的比值。如果测序深度的计算发生在序列比对前,定义为原始片段深度(raw read depth);如果在序列比对后计算测序深度,称为平均映射片段深度(mean mapped read depth)。⑤序列组装:与参考基因组序列进行比对分析,利用统计学计算,确定每个碱基位点的最大可能性的基因型,然后组装出整个基因组序列。
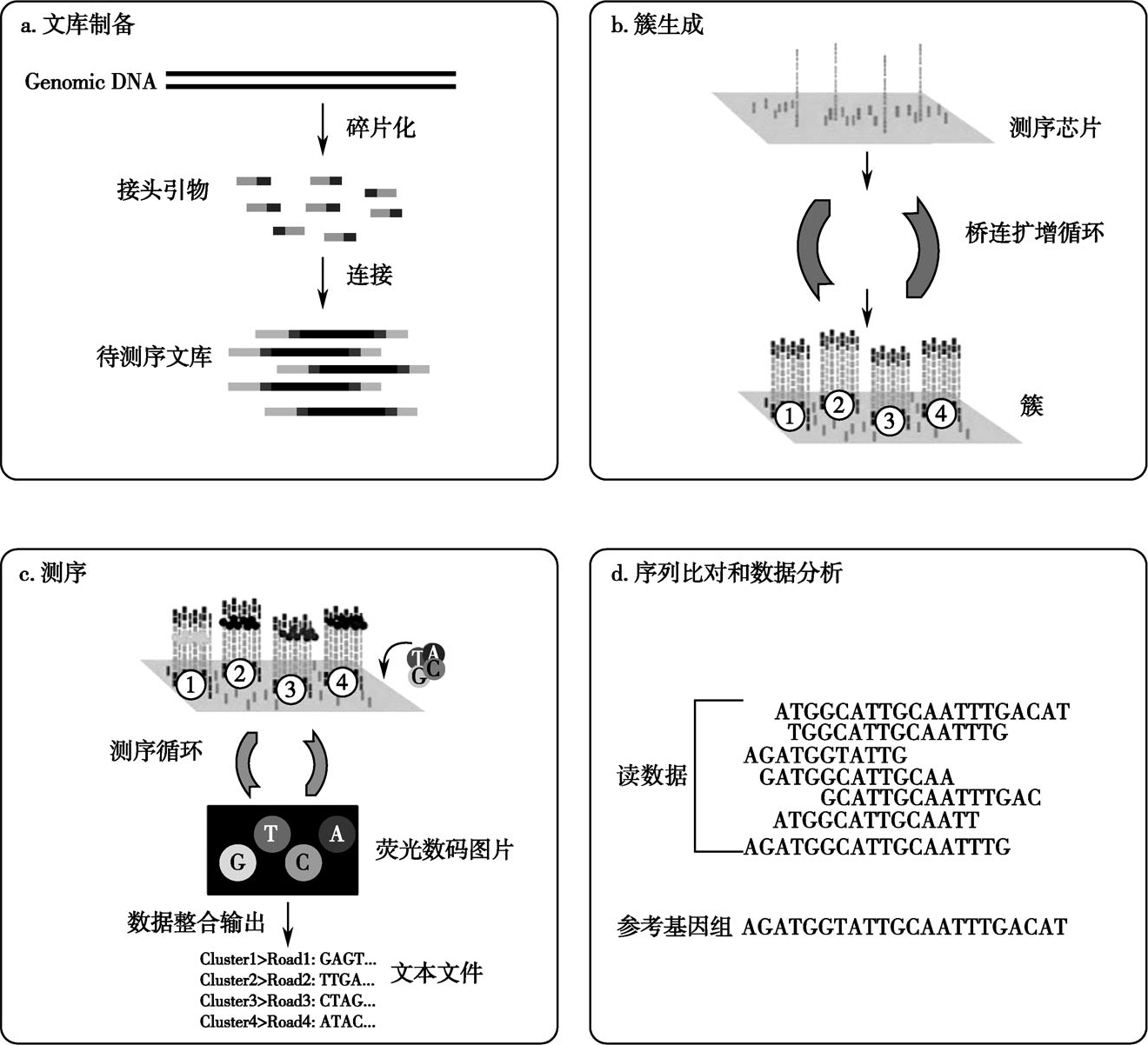
图2-5-4A 人全集因组测序示意
(Illumina的双末端测序方法的基本步骤引自https://www.illumina.com)

(资源11) 人全基因组测序示意(彩图)
3.全基因组重测序生物信息学分析(图2-5-4B)
(1)SNV与InDel的检测及注释:
基于序列比对和组装的结果,采用软件找到全基因组中所有的SNV和InDel,然后结合质量分值、测序深度、GC含量、重复度、杂合度等参数过滤并筛选出可信度高的SNV和InDel位点,并对所有SNV位点和InDel进行注释,包括各个位点对应的碱基与对应的氨基酸变化、可能引起的功能变化、分类以及在各个常用数据库中的标准代码格式等。
(2)结构变异检测及注释:
根据所得序列与参考基因组序列比对分析结果,检测全基因组中各种类型的结构变异,并对检测到的结构变异进行注释。常用的结构变异分析软件有CNVnator。
数据展示:用软件将分析结果通过图表和曲线形式,展示各种参数和突变分析结果。
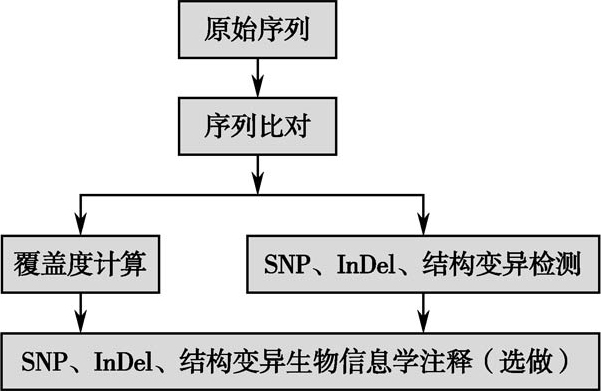
图2-5-4B 人全基因组测序生物信息学分析流程
二、人全外显子组与目标区域测序
(一)人全外显子组测序(exome sequencing)
常用目标序列捕获(target capture)技术,全基因组DNA中的全部外显子序列捕获并富集后,进行高通量测序分析。通常包括紧邻各外显子的5'端与3'端UTR的10bp。全外显子组的核苷酸对数只占全基因组1%~2%,因此全外显子组测序比全基因组测序覆盖度更深,数据准确性更高。目前估计DNA变异引起的疾病中,80%以上来自外显子组区域的变异。全外显子组测序特别适合于单基因和多基因遗传病的临床分析。
(二)人目标基因区域测序(targeted sequencing)
用目标序列捕获(target capture)技术,对相关蛋白质编码区域DNA或其他区域中某段特定序列捕获并富集后,进行高通量测序。只分析基因组中特定的疾病相关基因(gene panel),能够对指定目标区域进行深度测序,显著提高了对基因组中特定目标区域的检测灵敏度,并降低了检测成本。特别适用于肿瘤体细胞突变的检测。
(三)人目标序列捕获技术
主要分两类,探针杂交捕获(hybrid capture)和多重PCR捕获(multiplex PCR based target capture)。捕获得到目标序列,利用测序仪对目标序列进行测序分析。目标序列捕获技术与高通量测序技术结合,可以发现和分析各种类型的基因突变,帮助寻找复杂疾病的相关基因及突变位点。
1.探针杂交捕获
按目标基因区域序列,设计并制备特异性探针,与基因组DNA片段进行杂交,将目标基因DNA片段捕获并富集。探针可以锚定在芯片表面,也可呈液体状态,液相探针的方法更容易规模化,不需要处理芯片的特殊设备,价格便宜,特别适用于全外显子组和大基因测序。Thermal Fisher的全外显子测序的捕获流程如图2-5-5。
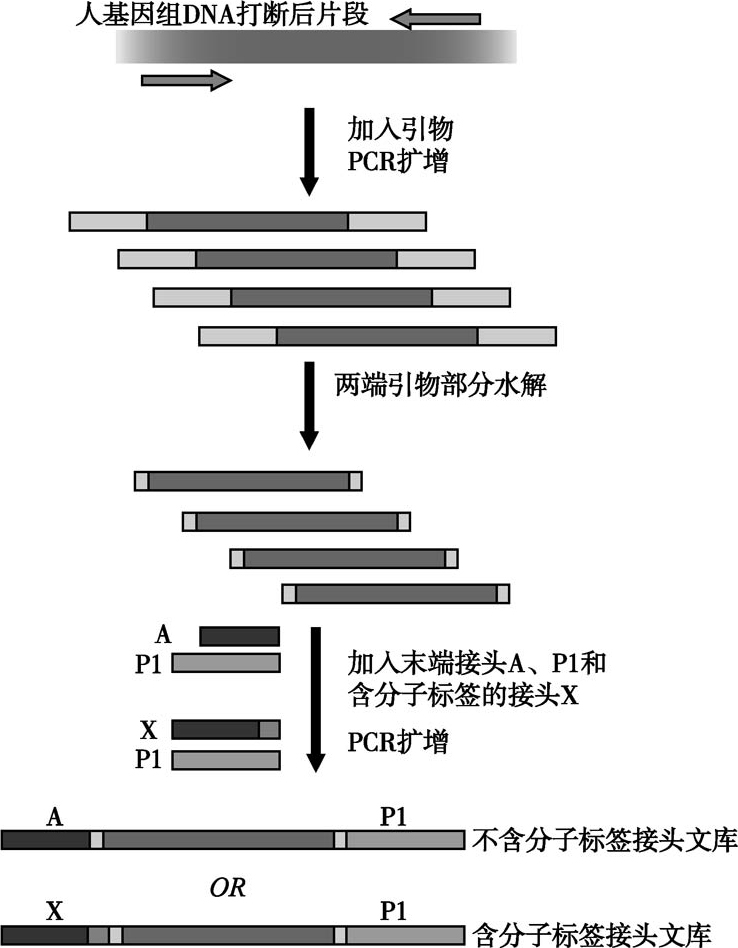
图2-5-5 目标序列捕获流程示意(Thermo Fisher AmpliSeq)
2.多重PCR捕获
①用一组已知限制性内切酶切断全基因组DNA;②DNA片段与生物素标记的探针在杂交缓冲液中杂交,然后DNA片段连接环化,环状DNA中加入能与测序引物结合的位点序列;③利用streptavidin标记的磁珠捕获并分离纯化杂交产物,除去未结合的DNA片段和DNA探针;④PCR扩增捕获的目标DNA片段并纯化PCR产物;⑤PCR产物经质量检测后,上测序仪检测序列。
目标基因多重PCR捕获法(multiplex PCR based target capture):适用于较小的外显子组,如5~50个基因的顺序。多重PCR捕获需要的样品量低(10ng),流程简单,成本低。
捕获样品转入Illumina测序仪测定核苷酸序列后与参考序列比对分析,获取样品核苷酸序列,找出突变位点和突变类型。
三、人全转录组测序
(一)全转录组测序(transcriptome sequencing)
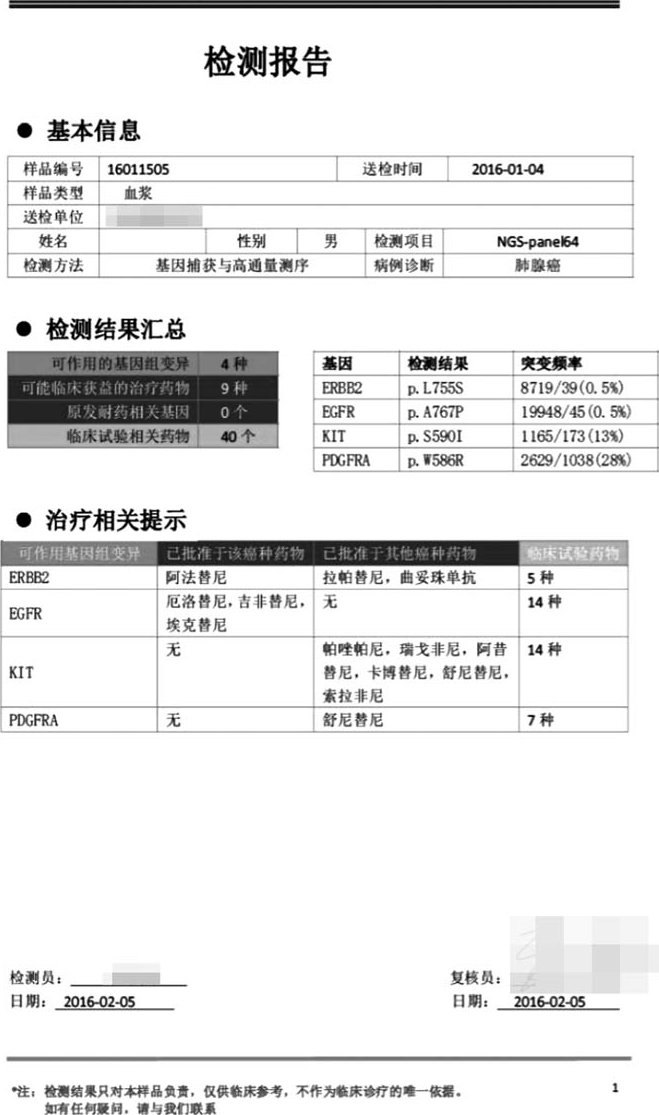
图2-5-6 临床肿瘤患者的基因捕获与高通量测序报告
同时测定特定细胞或组织在某一状态下所有的转录产物序列,分析基因结构与功能的关系、可变剪接位点和新转录产物预测等。与传统芯片杂交平台相比,高通量测序平台可以测定DNA序列未知转录组,提供整体转录活动更精确的定性定量信息。
(二)转录组测序的技术路线
以mRNA测序为例。
1.逆转录产物制备
提取样品总RNA后,用带有Oligo(d T)的磁珠富集mRNA。用化学方法将mRNA打断后作为模板,用六碱基随机引物(random hexamers)合成第一条cDNA。加入缓冲液、d NTPs、RNase H和DNA polymeraseⅠ,以第一条cDNA链为模板,合成第二条cDNA链,产物纯化后加接头,PCR扩增,末端修复后连接测序接头。
2.上机测序
详见“全基因组重测序的基本步骤”。
3.数据分析
根据参考基因组信息,将RNA-Seq测序所得数据做初步处理,与参考基因组比对,获得基因的表达信息,进行基因功能注释、基因结构分析、可变剪切分析、新基因的预测以及基因表达差异分析等。
四、检测报告
目前国内医院多数应用目标基因捕获高通量测序,全外显子组测序尚未推广。检测报告举例如图2-5-6,检测报告常附有用药建议和药物作用原理等。

